

SPECTROSCOPIE INFRAROUGE
Introduction
Les spectres infrarouges permettent une véritable étude fonctionnelle des molécules organiques.
Ce sont des spectres de vibration-rotation dans lesquels l’énergie de vibration est beaucoup plus grande que celle de la rotation.
Dans ce cours on étudiera l’aspect quantitatif de l’énergie de vibration-rotation, les instruments utilisés dans le domaine IR, l’interprétation des spectres et l’application de cette méthode.
– Le domaine spectral infrarouge

L’infra rouge analytique recouvre plusieurs méthodes d’identification non destructives basése sur l’absorption (ou la réflexion), par l’échantillon, des radiations électromagnétiques.
Cette bande spectrale est elle-même divisée en proche IR (de 0,7 à 2.5µm), en moyen IR (2.5-25 µm) et en lointain IR (au-delà).
Usuellement elle s’étend entre 600 et 4000 cm-1 certains appareil descendent jusqu’à 400 – 200 cm-1.
II-Origine de l’absorption lumineuse dans l’infrarouge
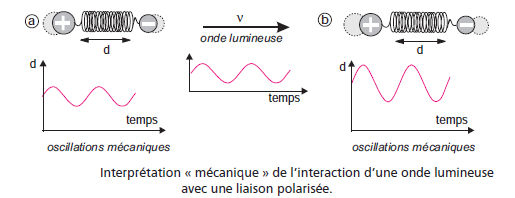
Dans le proche et le moyen infrarouge, l’absorption de la lumière par la matière a pour origine l’interaction entre les radiations de la source lumineuse et les liaisons chimiques animées d’un mouvement de vibration et de rotation et s’ils sont différents, ils forment un dipôle électrique oscillant à la même fréquence de l’onde qui irradie la liaison non symétrique et tournant autour d’un point fixe.
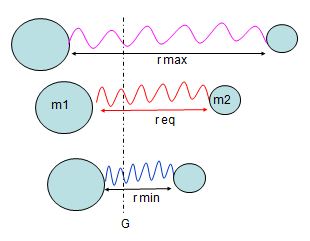
III-modèle simplifié des interactions vibrationnelles
Pour modéliser les vibrations des liaisons, on se réfère à l’oscillateur harmonique

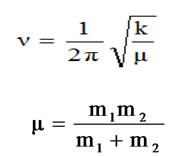
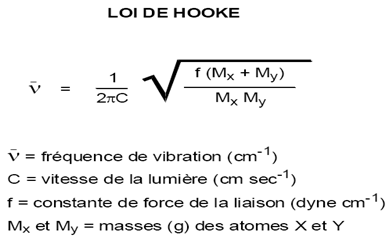
IV-Expression de l’énergie de vibration

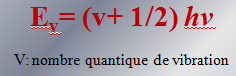
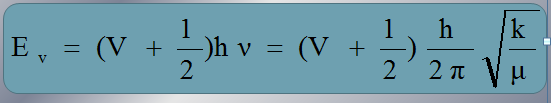
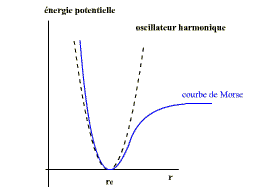
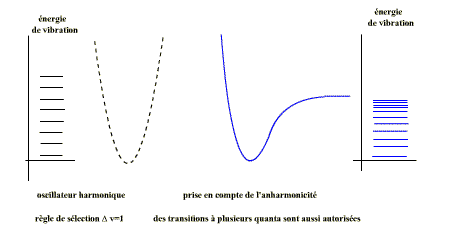
V-Cas réel de l’oscillateur anharmonique

L’assimilation à un oscillateur harmonique ne tient pas compte de la nature et de la complexité de la liaison chimique, et la courbe Ev=f(x0) n’est pas une parabole.
L’expression mathématique qui tient de la réalité est celle de Morse:
VI-Modes vibrationnels
1-Degrés de liberté:
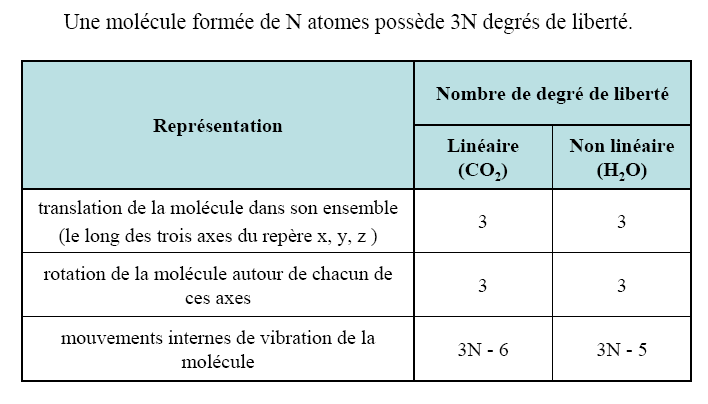
2-Modes vibrationnels: types simples

1-vibrations de valence ou d’élongation (symétriques ou antisymétriques): qui font intervenir une (des) variation(s) de(s) longueur(s) de liaison(s), les angles que forment ces liaisons restant constants, (stretching vibrations)
2- modes de déformation: pour lesquels, au contraire, les liaisons gardent leur longueur, mais les angles qu’elles forment varient (bending vibrations).Ces déformations touchent des molécules de 3 atomes et peuvent être dans ou hors du plan.
VII-Facteurs influents sur la vibration:
Vibration de valence
1-Effet de la force de la liaison:

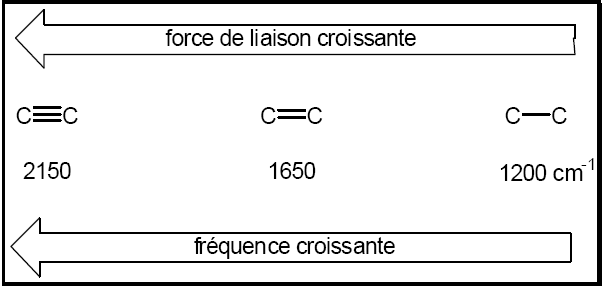
2-Effet de la masse des atomes :
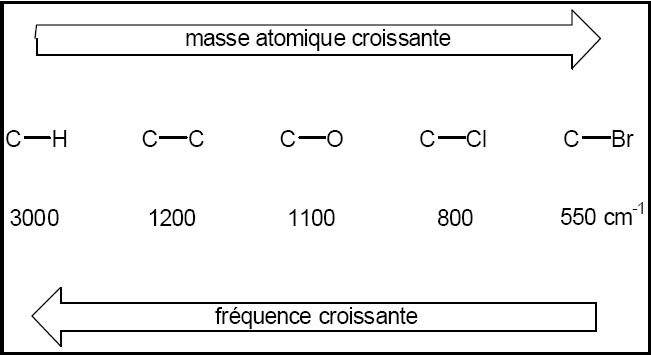
3-Effet de la polarisation du lien:
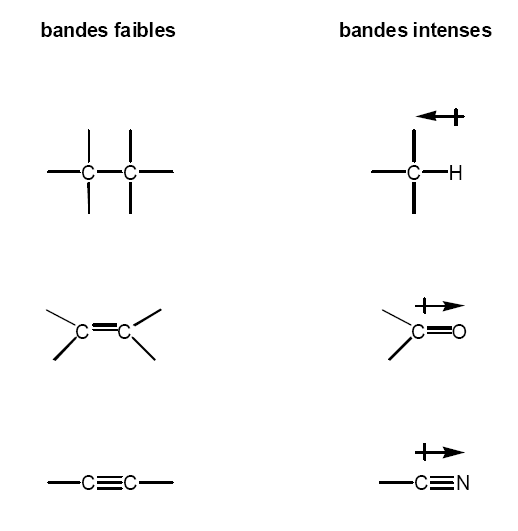
Seules les vibrations impliquant une variation du moment dipolaire de la molécule s’observent en infrarouge.
Conséquemment, la vibration de liens polarisés donnera lieu à des bandes intenses, alors que les bandes de liens non-polarisés seront peu ou pas visibles.
4-Effet de la liaison hydrogène:
La liaison hydrogène réalisée entre un hydroxyle et un carbonyle entraine le déplacement de vibration de OH à ν =3000cm1- ou même moins.

5-Effet de la conjugaison:
La conjugaison entraine une délocalisation des e– П, la force de rappel est donc affaiblie par rapport à une double liaison et entraine des diminutions de ν .
Vibration de déformation


-Vibrations nombreuses beaucoup plus sensibles à l’environnement que celles de valence, car elles ont besoin pour se produire d’un volume plus important et risquent d’être entravées par la présence d’atomes voisins
Origine des bandes PIR: harmoniques et bandes de combinaison
Des interactions entre les différents modes de vibration peuvent donner naissance à des bandes de combinaisons qui apparaissent à des fréquences qui sont des combinaisons linéaires des bandes fondamentales : v = av1 + bv2 ( a et b sont des nombres entiers.)
Les bandes de combinaisons+ les harmoniques:
-Fréquences supérieures à celles des bandes fondamentales qui leur ont donné naissance
– Bande plus large et de plus faible intensité que les bandes fondamentales
VIII-Modèle simplifié de la rotation moléculaire


IX-Conditions de transitions: spectre de rotation-vibration

– ∆ E= hv
-P variable au cours du mouvement
-∆V=+1 et ∆ J= ±1 à satisfaire en même temps
X-Instrumentation
a)Analyseur faisceau simple, comportant un monochromateur fixe ou un filtre, utilisé lorsqu’une mesure à longueur d’onde unique suffit ;
b) spectromètre double faisceau de type dispersif.
Contrairement aux spectrophotomètres de l’UV/Visible, l’échantillon, placé avant le monochromateur, est soumis en permanence à tout le rayonnement de la source.
Cette catégorie utilise des filtres ou un monochromateur installés dans des montages particuliers en fonction de la plage spectrale étudiée.
c) modèle simple faisceau à transformée de Fourier.
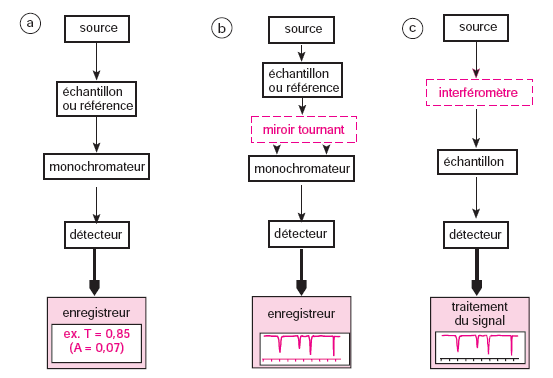
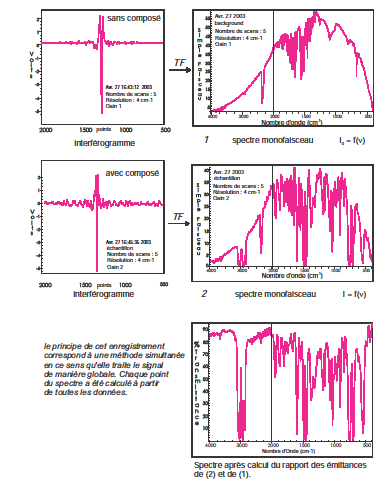
Modèle simple faisceau à transformée de Fourier:
Les spectromètres infrarouges à transformée de Fourier correspondent à un montage optique à simple faisceau qui comporte comme pièce essentielle un interféromètre (type Michelson ) placé entre la source et l’échantillon.
L’ensemble des interférences positives et négatives produit un interférogramme qui contient toutes les informations requises pour produire un spectre, suite à une opération mathématique appelée transformée de Fourier
Sources lumineuses
-Gros filament ou bâtonnet creux porté à l’incandescence
-Mélange d’oxydes de zirconium et de terres rares : source de Nerst
-carbure de silicium: lampe Globart
-lampe halogène-tungstène (PIR)
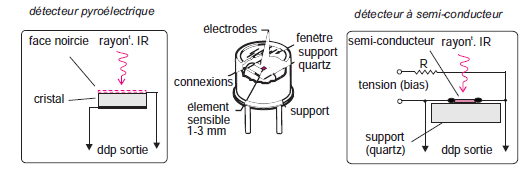
les détecteurs:
-La détection repose sur l’effet thermique des radiations.
-Thermistors, thermocouples, thermopiles
-Pour les spectromètres à transformée de Fourier: cristal pyroélectrique ou un semi-conducteur du type photodiode
Préparation des échantillons:
– Les plus courants: NaCl,KBr (fragiles et solubles dans l’eau),CsI (transparent jusqu’à 200 cm−1), AgCl, le KRS-5 (bromoiodure de thallium) ,Le diamant ( durs et insolubles et plus chers.), les AMTIR verres composés de germanium, d’arsenic et de sélénium (ex.
Ge33As12Se55),Polyéthylène (carbure saturé, peu de bandes d’absorption, inerte vis-à-vis de l’eau: dosage en solutions aqueuses)
Procédés par réflexion pour les échantillons solides
Accessoire de Réflexion Totale Atténuée (ATR).
– Plusieurs réflexions du faisceau optique à l’interface entre l’échantillon et un matériau transparent d’indice de réfraction n élevé tel le diamant (n = 2,4) sur lequel il a été déposé.
-la lumière pénètre faiblement dans l’échantillon (quelques dixièmes de micromètre) (onde évanescente).
– Succession de plusieurs réflexions
Totales mais atténuées (trajet optique
Effectif comparable à celui qui aurait
Été obtenu par transmission)
–On corrige néanmoins le spectre pour
tenir compte de la profondeur de pénétration.
Procédés par transmission
–Les gaz:
Cuves en sel gemme ou cellules pour lesquelles le trajet optique peut être très grand (les absorbances sont faibles)
–Les liquides:
-cellules à film entre disques de NaCl ou de KBr (a.quant)
-cuves en quartz ou en matériaux conventionnels (a.qualit)
-Les solides :
-En solution, -Réduit en poudre fine
( huile de paraffine, KBr anhydre)

XI-Interprétation des spectres
1- Un groupe CARBONYLE est-il présent?
Le groupe C=O génère une bande intense dans la région 1820 et 1660 cm-1.
● Quel type???
ACIDE: un groupe OH est-il aussi présent? Recherchez une bande large vers 3400-2400 cm-1.
AMIDE: un groupe NH est-il aussi présent? Recherchez une bande d’intensité moyenne vers 3500 cm-1.
Si présent, le pic est-il simple (NH) ou double (NH2)?
XII-Applications de la spectroscopie IR
1-L’analyse qualitative:
-Tentative d’identification du composé analysé par comparaison à des spectrothèques générales ou spécialisées
-Le spectre à comparer est mis en conformité (absorbance et longueurs d’onde) avec le modèle des spectres en bibliothèque.
-La recherche : comparaison mathématique du spectre du composé inconnu avec tous ceux qui forment la spectrothèque considérée (étape est difficile à cause de la complexité du spectre.)
-La tentative d’attribution de la majorité des bandes caractéristiques du spectre est parfois très abordable surtout si la formule brute du composé est connue
4-Des TRIPLES LIAISONS sont-elles présentes?
Les NITRILES ont une bande fine d’intensité moyenne (C≡N) vers 2250 cm-1.
Les ALCYNES ont une bande fine de faible intensité (C≡C) vers 2150 cm-1.
Recherchez aussi la présence de la bande ≡C-H vers 3300 cm-1 afin de déterminer si l’alcyne est terminal ou pas.
5-Le groupe NITRO est-il présent?
Recherchez la présence de deux bandes NO2 intenses vers 1600-1500 cm-1 et 1390-1300 cm-1.
Si votre analyse n’a révélé la présence d’aucun de ces groupes fonctionnels vous avez probablement un ALCANE.
Vous devriez avoir un spectre assez simple avec des bandes CH à la droite de 3000 cm-1.
2-Si C=O est absent, recherchez la présence des fonctions suivantes:
ALCOOL: recherchez la large bande OH vers 3600-3300 cm-1.
PHÉNOL: pour les phénols, confirmez aussi la présence d’un cycle aromatique
2-Des DOUBLES LIAISONS ou des CYCLES AROMATIQUES sont-ils présents?
Les liens C=C génèrent une bande faible vers 1650 cm-1.
Des bandes, moyennes à fortes, dans la région de 1650 à 1450 cm-1 indiquent souvent la présence d’un cycle aromatique.
La présence de bandes CH à la gauche de 3000 cm-1 (=C-H) confirme la
présence d’une ou plusieurs insaturations.
ESTER: un lien C-O est-il présent? Recherchez une bande intense vers 1300- 1000 cm-1.
ANHYDRIDE: y-a-t-il deux bandes carbonyles (vers 1810 et 1760 cm-1), plutôt qu’une seule?
ALDÉHYDE: les deux bandes CH caractéristiques d’un aldéhyde sont-elles présentes vers 2850 et 2750 cm-1 (i.e. à la droite des autres bandes CH)?
CÉTONES: vous avez une cétone si les cinq autres options ont été éliminées.
Conclusion
Cette méthode d’analyse est simple à mettre en œuvre et non destructrice.
Elle permet d’analyser aussi bien les matériaux organiques que les matériaux inorganiques.
La grande diversité des montages expérimentaux permet la caractérisation de pratiquement tout type d’échantillon, quel que soit leur état physique ou de surface, cependant pour améliorer encore notre analyse d’autres techniques complémentaires sont utilisées comme RMN
AVANTAGES
– Peu ou pas de préparation de l’échantillon
– Analyse rapide en temps réel -Coût de l’analyse peu élevé
– Méthode de choix pour le contrôle industriel, analyse in situ en temps réel
INCONVENIENTS
– Manque de corrélation structurale (difficultés pour l’interprétation des spectres)
– Besoin de calibration pour les mélanges (analyse directe très difficile en général)
-Utilisation des méthodes de chimiométrie: Phase d’étalonnage longue et délicate
Place du PIR en analyse pharmaceutique :
1-Contrôles sur les matières premières:
-Identification des matières premières
-Qualification de principe actif: Polymorphisme et cristallisation, Discrimination d’aromes, Contrôle de granulométrie
2-Contrôle en cours de fabrication de l’homogénéité d’un mélange, l’épaisseur d’un pelliculage
3-Produit fini: Uniformité de teneur, Lyophilisats: teneur en eau
2-2-Analyse quantitative dans le proche infrarouge
-Essentiellement réservé aux analyses de contrôle
– On ne peut que très rarement utiliser une méthode d’étalonnage basée sur l’absorbance à une seule longueur d’onde comme en spectrométrie UV–VIS cependant Le passage par les courbes dérivées améliore la précision (spectres bruts sont assez souvent d’aspect déroutant par leur absence de bandes d’absorbance nettes)
-Les mesures sont effectuées dans des cuves de 1 cm de parcours optique, et sur le composé sans dilution (Les bandes d’absorption sont peu intenses.) et on opère en transmission ou en réflexion
– La grande majorité des applications repose sur des méthodes chimiométriques permettant la prévision de variables quantitatives
2-L’analyse quantitative
2-1-Analyse quantitative dans le moyen infrarouge:
-Echantillons solides dispersés au sein d’un disque de KBr : on ajoute un composé à usage de référence interne (carbonate de calcium, naphtalène, nitrure de sodium), en égale quantité à tous les standards ainsi qu’à l’échantillon.
-Echantillons liquides: mesures d’absorbance dans des cuves à parcours optique faible, pour minimiser l’absorbance propre au solvant, dont aucun n’est vraiment transparent dans ce domaine.