

Pharmacologie des antihistaminiques
Introduction
L’histamine est un médiateur qui intervient dans la sécrétion gastrique, la régulation de la vigilance et certaines manifestations allergiques.
Domaine histaminergique
Localisation et libération de l’histamine
Localisation de l’histamine
L’histamine est présente dans les mastocytes tissulaires et les granulocytes basophiles du sang.
L’histamine des mastocytes représente la réserve stable et lentement régénérée.
Les mastocytes sont répartis dans la peau, l’intestin, le foie, les bronches, les tumeurs.
L’histamine peut également être synthétisée (mais non stockée) dans les plaquettes, les cellules dendritiques, les lymphocytes, ainsi que dans les cellules pariétales et principales de l’estomac.
Elle est enfin formée au niveau des neurones cérébraux, d’où elle peut être rapidement libérée et régénérée.
Libération de l’histamine
Le mécanisme physiopathologique principal de la libération d’histamine est de type immunologique.
Cette libération peut aussi survenir sous l’influence de phénomènes physiques, tels que l’irritation de la peau, l’exposition au soleil ou à des radiations, ou lors de variations de température ou de pression.
L’histamine est enfin libérée sous l’action de nombreux facteurs chimiques : venins, toxines, médicaments (morphine, codéine, pentamidine, tubocurarine, guanéthidine, mépéridine…), ou de réactifs pharmacologiques.
Métabolisme de l’histamine
Structure et synthèse
L’histamine, nom usuel de la 2-(4-imidazolyl) éthylamine, est une monoamine primaire.
Fig. 1.
Structure de l’histamine
L’histamine est obtenue en éliminant le groupe carboxylique de l’histidine.
De nombreux tissus animaux (mastocytes, muqueuse gastrique…) contiennent de
l’histidine et l’enzyme (histidine décarboxylase) qui catalyse la formation de l’histamine.
Dans certaines cellules, l’histamine est stockée sous forme de granules.
Catabolisme
L’histamine est rapidement inactivée par :
Méthylation en N-1-méthyl-histamine (70 %) sous l’influence de l’histamine-N- méthyltransférase (HNMT) ;
Oxydation de la chaîne latérale par une diamino-oxydase (histaminase) qui permet la formation de l’acide imidazolacétique.
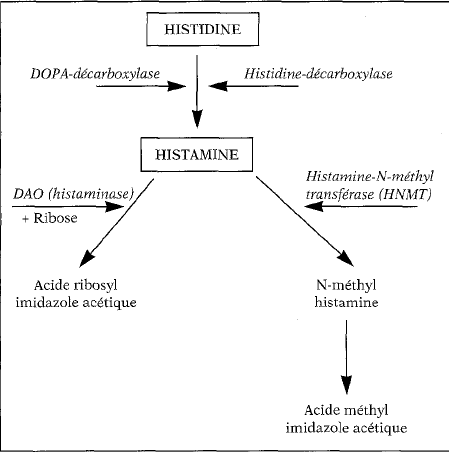
Fig.2 Métabolisme de l’histamine
Effets et récepteurs
Il existe au moins deux types de récepteurs histaminiques postsynaptiques appelés H1 et H2, des récepteurs H3 surtout présynaptiques, présents notamment dans le cerveau, et des récepteurs H4 décrits plus récemment.
Le rôle des récepteurs H1 et H2 est le mieux connu.
Effets H1 : La stimulation des récepteurs H1 entraîne :
Contraction des fibres lisses, notamment bronchiques et digestives.
une vasodilatation capillaire.
par effet central, une augmentation de la vigilance
Effets H2 : La stimulation des récepteurs H2 qui agissent par l’intermédiaire de l’AMP cyclique entraîne :
Une augmentation de la sécrétion gastrique d’acide chlorhydrique qui peut être considérée comme le principal effet H2.
une stimulation cardiaque : effets inotrope et chronotrope positifs.
une vasodilatation :
La stimulation par l’histamine des autocécepteurs présynaptiques H3 réduit la libération d’histamine au niveau du système nerveux central et périphérique et le blocage de ces récepteurs augmente la libération d’histamine qui stimule la vigilance.
Pharmacologie des antihistaminiques H1
Mode d’action et propriétés pharmacologiques
Les anti-H1 sont des agonistes inverses des récepteurs H1.ils vont donc s’opposer aux effets d’activation du récepteur H1 par l’histamine notamment sur la peau ,les vaisseaux et les muqueuses conjonctivales,nasales,bronchiques et intestinales.
Ils inhibent ainsi les effets H1 de l’histamine et plus particulièrement l’effet vasodilatateur et l’augmentation de la perméabilité capillaire à l’origine des réactions œdémateuses.
Certains composés manquent de sélectivité notamment les anti-H1 de première génération et sont aussi des antagonistes compétitifs des récepteurs muscariniques de l’acétylcholine, leur conférant des propriétés anticholinergiques.
Dans une moindre mesure, certains anti-H1 vont inhiber les récepteurs adrénergiques et sérotoninergiques.
Les anti-H1 de première génération, dits anticholinergiques, comme la dexchlorphéniramine,la prométhazine ou l’hydroxyzine.
En général, ils sont capables de traverser barrière hémato-encéphaliques et sont donc sédatifs (sauf la méquitazine), et ils présentent également une action antiémétisante par inhibition de la zone chimio-sensible.
Les anti-H1 de deuxième génération, non anticholinergiques comme la loratadine ou la cétirizine sont le plus souvent non sédatifs du fait de leur faible pénétration dans le cerveau.
Ils n’ont pas en principe d’effet sur la repolarisation cardiaque.
Pharmacocinétique
La plupart des anti-H1 sont bien absorbés par voie orale, avec une bonne biodisponibilité.
Le métabolisme des anti-H1 est très variable allant de pas ou peu de métabolisme (féxofenadine) à un métabolisme total (via les cyp 450) produisant des composés dont
certains sont pharmacologiquement plus actifs de la molécule mère. (Ex : la desloratadine est un métabolite actif de la loratadine.
L’élimination est également variable (urinaire ou fécale).
Les anti-H1 de deuxième génération ont pour la plupart une longue durée d’action soit en raison d’une longue demi-vie, soit en raison de la formation de métabolite(s) actif(s).
Pour ces molécules, une seule administration quotidienne est suffisante.
Indications
Les antihistaminiques H1 sont utilisés pour le traitement symptomatique de diverses manifestations allergiques cutanées (urticaire) ou muqueuses (rhinite, rhume des foins, conjonctivite).
Ils ne sont pas efficaces dans l’asthme.
Autres indications
Sédatif, anxiolytique (hydroxyzine Atarax®) voire hypnotique (doxylamine Donormyl ®).
Antitussif, toux non productives allergiques et irritatives (alimémazine Théralene®, oxymémazine Topléxil®, chlorphénamine Humex®).
Antiémétique dans le mal des transports avec participation de l’effet cholinergique (dimenhydrate Nausicalm®, diphenhydramine Nautamine®).
Prémédication avant une anesthésie générale ou un traitement allergisant (chimiothérapie par exemple).
Effets indésirables
Effets dépresseurs centraux (sédation).
Effets anticholinergues ; essentiellement pour les molécules de première génération.
Effets cardiaques ; troubles de rythme.
Réactions allergiques ; des réactions allergiques paradoxales peuvent survenir en particulier pour les formes topiques.
Grossesse et allaitement
En raison d’effets tératogènes observés chez l’animal avec certains antihistaminiques, la plupart des molécules sont contre-indiqués chez la femme enceinte.
L’utilisation d’anti H1 administrés par voie locale est envisageable en raison d’un moindre passage systémique.
La majorité des anti-H1 passe dans le lait maternel, compte tenu des possibilités de sédation ou d’excitation paradoxale du nouveau-né, ces médicaments sont déconseillés en cas d’allaitement.
Compte tenu des données disponibles, l’utilisation de la doxylamine est possible au cours de la grossesse quel qu’en soit le terme.
Elle est même indiquée dans certains pays (Canada, USA.
Royaume Uni) pour traiter les nausées et vomissements de la grossesse.
Contre-indications
Liées aux effets dépresseurs centraux : conduite automobile.
Liées aux effets anticholinergiques (molécules de première génération) :
Glaucome par fermeture de l’angle.
Rétention urinaire.
Liées au risque de troubles du rythme cardiaque de certaines molécules avec les arythmies cardiaques et les cardiopathies.
Pharmacologie des antihistaminiques H2
Les antagonistes H2 ou antihistaminiques H2 ont été découverts beaucoup plus tardivement que les antagonistes H1.
Le premier médicament commercialisé a été la cimétidine suivie de la ranitidine, la famotidine et la nizatidine.
Rappel physiologique
Sous l’action de la gastrine, l’histamine est un médiateur libéré par les cellules entérochromafines qui favorise le relargage d’acide chlorhydrique dans l’estomac par la pompe à protons, en stimulant ses récepteurs H2 localisés au pôle basal des cellules pariétales gastriques.
Mode d’action des anti-H2
Les anti-H2 sont des antagonistes des récepteurs H2 réduisant cette sécrétion acide.
L’action des anti-H2 est donc limitée à une partie des mécanismes de stimulation de la sécrétion acide gastrique.
Pharmacocinétique
L’absorption après administration orale est rapide, La biodisponibilité varie en fonction de la molécule.
Leur élimination est essentiellement rénale.
La cimétidine possède une particularité : alors que les autres antagonistes H2 ont peu d’effet sur le cytochrome P-450, la cimétidine l’inhibe et peut provoquer des interactions avec beaucoup de médicaments.
Indications
Comme l’acidité gastrique favorise l’apparition des ulcères et retarde leur cicatrisation, les antihistaminiques H2 sont indiqués dans le traitement des ulcères œsophagiens (par reflux acide) et des ulcères gastriques et duodénaux ainsi que dans celui des hémorragies digestives hautes d’origine ulcéreuse où on recourt surtout aux antagonistes H2 injectables.
Effets indésirables
Certains effets sont rapportés avec tous les anti-H2 : diarrhée, asthénie, douleurs musculaires, éruptions cutanées.
Une élévation des transaminases et de la créatininémie sont également possibles.
Tous les antagonistes H2 peuvent donner une bradycardie par suppression de l’effet chronotrope positif de l’histamine généralement sans gravité.
La cimétidine a des effets indésirables de type endocrinien : elle augmente la concentration de prolactine plasmatique et peut entraîner une gynécomastie et une galactorrhée.
Grossesse et allaitement
L’utilisation des anti-H2 est déconseillée en cas de grossesse et allaitement.
NB.
Insuffisance rénale et ou hépatocellulaire : il sera conseillé de réduire la posologie et l’adapter en fonction de la créance a la créatine.
Remarque : Le seul agoniste histaminergique commercialisé aujourd’hui est la bétahistine Serc® qui est un faible agoniste H1 utilisé pour ses propriétés vasodilatatrices dans le traitement des syndromes de Ménière (vertiges, acouphènes, surdité).