
LES METHODES EN ANALYSE IMMEDIATE
INTRODUCTION :
L’analyse chimique est l’ensemble de procédures permettant l’identification (analyse qualitative pour la détermination de la nature) et la quantification (dosage quantitatif) de la composition d’un échantillon.
L’analyse chimique immédiate est la séparation des corps purs d’un mélange.
Un procédé de séparation est une technique permettant de transformer un mélange de substances en deux ou plusieurs composants distincts.
Ses buts peuvent être divers ; la purification, la concentration, le fractionnement.
DEFINITIONS :
Un mélange est une association de deux ou plusieurs substances qui n’interagissent pas chimiquement.
Il peut être hétérogène ou homogène.
Un mélange hétérogène (au moins deux phases) est formé de plusieurs corps visibles les uns par rapport aux autres.
Il peut être obtenu par suspension d’un solide dans un liquide ou par mélange de deux liquides non miscibles.
Un mélange homogène comprend une seule phase formée de plusieurs corps que l’on ne peut pas distinguer à l’œil nu.
Il peut être obtenu par dissolution d’un solide ou un gaz dans un liquide ou par mélange de deux liquides miscibles.
CLASSIFICATION :
Les composants d’un mélange retiennent leurs propriétés physicochimiques propres.
Pour séparer ces composants, on met à profit certaines différences de propriétés entre le composé d’intérêt et le reste du mélange (états physiques, densité, températures de changement d’état).
Plus cette différence sera grande, plus la séparation sera aisée.
Ainsi, Le choix de la technique appropriée varie en fonction du mélange (état physique et type), de la substance que l’on doit séparer et des phases qui constituent le mélange.
Ce qui nécessite une bonne connaissance de la composition du mélange et des propriétés individuelles des différents composants.
On distingue deux grandes classes de procédés de séparation :
Les techniques physiques :
Ces techniques se basent sur des propriétés physiques mécaniques ou des phénomènes de diffusion.
La séparation mécanique est basée sur ; la mouillabilité de surface (flottation), la masse volumique (sédimentation, décantation, centrifugation, essorage), la granulométrie
(filtration, tamisage, chromatographie d’exclusion stérique), la mobilité électrique (électrophorèse), la mobilité magnétique (séparation magnétique des métaux ferreux).
La Séparations par diffusion englobe les techniques chromatographiques, les extractions, les séparations thermiques basées sur les transitions de phase (évaporation, distillation, cristallisation, sublimation), les séparations membranaires par diffusion (osmose et osmose inverse, dialyse, pervaporation) et les séparations par transfert d’ions (échange d’ions, techniques électromembranaires).
Tableau. 1. Le principe de séparation des principales méthodes de séparation
| Méthodes de séparation | Principe |
| Tamisage, Filtration | Taille des particules |
| Décantation, Sédimentation | Grande différence de densité |
| Centrifugation | Faible différence de densité |
| Évaporation, Distillation simple | Grande différence entre les points d’ébullition |
| Distillation fractionnée | Faible différence entre les points d’ébullition |
| Sublimation | Différence entre les points de sublimation |
| Cristallisation, précipitation | Solubilité, point de solidification |
| Chromatographie, Extraction | Différence de polarité, Solubilité |
Les techniques chimiques :
Il existe des techniques plus complexes de séparation des mélanges, qui nécessitent l’ajout de réactifs pour initier une réaction chimique (précipitation).
METHODES DE RESOLUTION DES MELANGES HETEROGENES :
Les mélanges hétérogènes sont faciles à séparer.
Il existe de nombreuses techniques.
Le choix de la technique dépend de la nature de mélange hétérogène.
Pour un mélange solide – solide, on peut utiliser plusieurs procèdes dont le tri (à la main, à la pince, magnétique) et le tamisage.
Un mélange solide – liquide peut être séparé par sédimentation, filtration, centrifugation.
Deux liquides non miscibles sont facilement séparés par décantation.
La décantation :
C’est un procédé de séparation mécanique qui permet de séparer les constituants d’un mélange hétérogène composé de deux liquides non miscibles sous l’effet de leur densité ou un solide en suspension d’un liquide sous l’effet du poids (souvent nommé sédimentation).
La séparation se réalise en laissant seulement se reposer le mélange.
Les corps les plus lourds vont alors se déposer dans le fond du récipient.
Lorsque les deux phases sont bien distinctes, on peut les séparer facilement.
Cette méthode simple n’est pas couteuse car elle nécessite peu de matériel mais son temps de réalisation est long et elle n’est pas sélective.
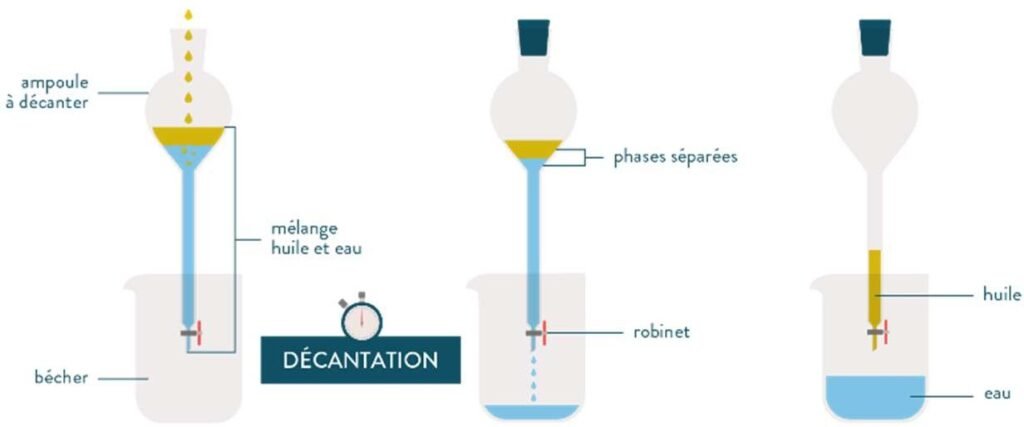
Figure.2. La décantation de deux liquides non miscibles
Pour un mélange solide-liquide, le dépôt solide est appelé sédiment (voir figure.3.).
La séparation obtenue est parfois partielle ne permettant pas d’obtenir un liquide limpide.
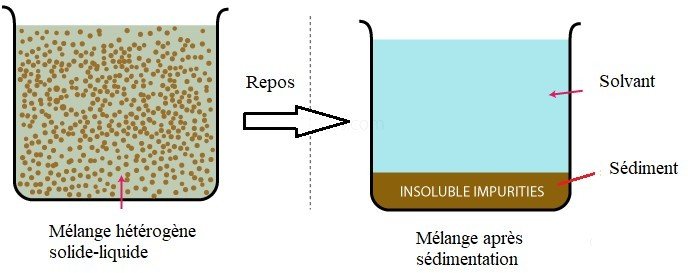
Figure.3. La sédimentation d’un mélange hétérogène solide liquide
La filtration
La filtration est une technique qui permet de séparer physiquement les constituants d’un mélange lorsqu’un des constituants est sous la phase liquide ou gazeuse et l’autre, sous la phase solide qui y est insoluble.
Pour ce faire, on utilise une substance poreuse minérale ou organique (voir figure.5.), appelée filtre.
Ce dernier doit être chimiquement inerte et il permet de retenir les particules solides en suspension dans le mélange qui sont plus grosses que ses pores (voir tableau.2.).
Le mélange à filtrer est versé progressivement, le liquide limpide qui passe au travers est appelé filtrat (il ne contient plus de particules solides).
Le solide que l’on recueille dans le filtre est appelé résidu, gâteau ou rétentat.
Cette méthode est rapide et efficace si le filtre choisi est adapté au mélange filtré.
Elle est souvent utilisée en complément d’une décantation, qui permet d’éliminer de nombreuses particules et évite au filtre de se boucher pendant la filtration.
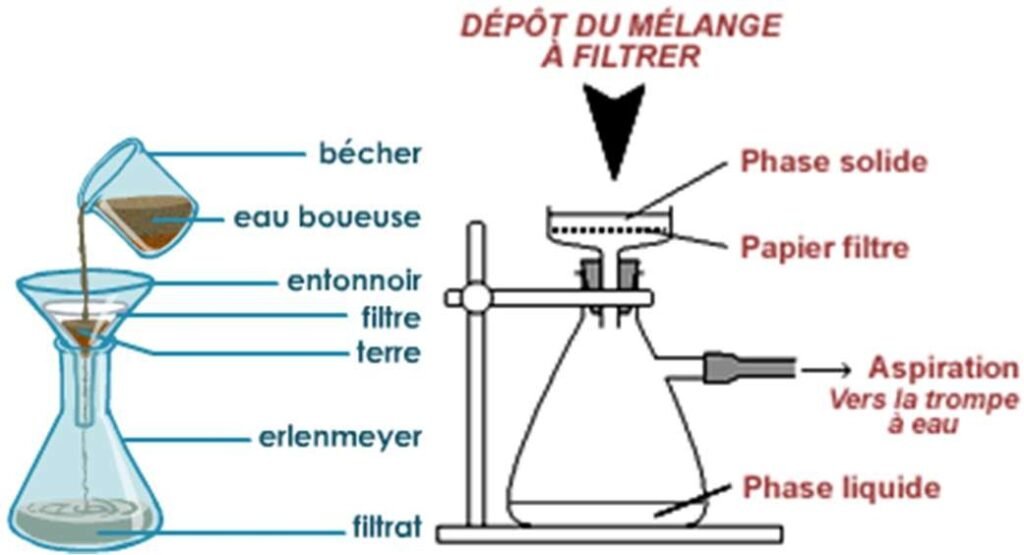
Figure.4. Procédure de filtration à l’aide d’un papier filtre à pression atmosphérique (à gauche), sous vide (à droite)
Elle peut s’effectuer sous, pression atmosphérique (par gravité), pression réduite (par aspiration), à chaud ou à froid.
La filtration à pression atmosphérique est lente contrairement à la filtration sous vide qui utilise un montage pour réduire la pression et accélérer l’opération.
Les filtres
Organiques
Minérales
-Dérivés de la silice
Papiers filtres Pates de cellulose Polymères
Figure.5. Les différents types des filtres
Le filtre le plus utilisé est en papier, qui doit être placé dans un entonnoir ordinaire, BUCHNER ou HIRSCH.
Il existe différents type de papier caractérisé par la taille des pores, par sa rétention de liquide et sa vitesse de filtration.
Les papiers filtres ronds doivent être
préalablement pliés pour leur donner une forme conique.
En fonction de la taille des particules retenues, on distingue différentes type de filtration (voir tableau.2.).
Tableau.2. Les différentes porosités du filtre en verre fritté
| Porosité | Taille des pores (µm) | Utilisation |
| 0 | 160-250 | gaz, gros précipités |
| 1 | 100 – 160 | gaz, précipités gélatineux |
| 2 | 40 – 100 | précipités cristallins, Hg |
| 3 | 16 -40 | précipités mi-fins (microfiltration) |
| 4 | 10 – 16 | précipités très fins (ultrafiltration) |
| 5 | 1 – 16 | filtration bactérienne (stérilisante) |
Filtration clarifiant
La centrifugation
Lorsque la décantation de particules sous l’effet du poids est inefficace (particules légères) ou trop lente, on a alors recours au procédé de centrifugation qui permet de séparer de deux à trois phases par l’action de la force centrifuge.
Pour cela, on substitue au champ de pesanteur terrestre un champ de forces centrifuges infiniment plus grand.
Le mélange est entraîné dans un mouvement de rotation très rapide.
Les particules solides les plus lourdes sont alors poussées vers les parois du récipient (culot), alors que les particules les plus légères et les liquides restent en surface, ce que l’on nomme surnageant.
L’appareil utilisé, appelé centrifugeuse, est constituée d’un axe de rotation enfermé dans une chambre de centrifugation.
Il s’agit d’un appareil doté de tubes destinés à contenir des mélanges et pouvant tourner autour d’un axe : plus la rotation est rapide, plus la centrifugation est efficace.
Les échantillons à centrifuger doivent être équilibrés deux à deux symétriquement placés par rapport à l’axe de rotation.
Elle permet d’obtenir les mêmes résultats qu’une décantation, mais plus rapidement.
Il y’a plusieurs types de centrifugeuses dont les centrifugeuses analytiques et/ou préparatoires, les ultracentrifugeuses et micro- centrifugeuses.
Le choix des centrifugeuses repose sur la vitesse de rotation, le volume de l’échantillon et le type de rotor.
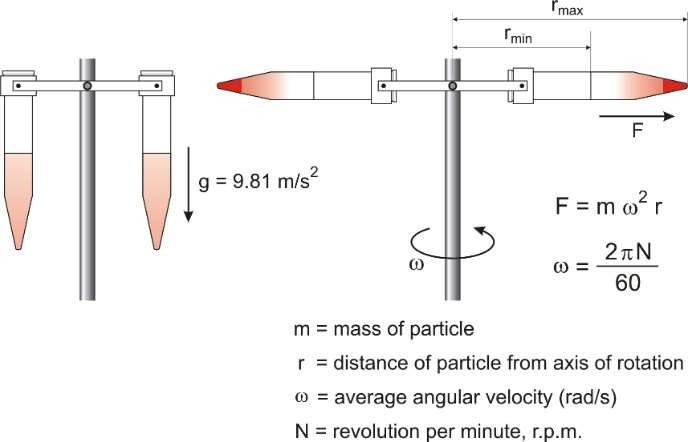
Figure.6. Procédure de centrifugation
Applications :
La décantation, la filtration et la centrifugation sont des procédés permettant de préparer des échantillons avant analyse.
Sachant que, suivant les cas, on cherche à récupérer : soit la phase continue débarrassée au maximum des matières en suspension (dans le cas, par exemple, d’analyse d’air, d’eau, d’un liquide alimentaire…) ; soit la phase dispersée (analyse de précipités de cristaux…).
Puisque toutes ces méthodes manquent de sélectivité, elles doivent être complétées par des méthodes plus sélectives (méthodes d’extraction liquide-liquide ou les méthodes chromatographiques…etc).
En pharmacie, on utilise les méthodes de filtration et centrifugation pour la purification des solutions, dans la préparation des solutions stériles ou les échantillons analytiques.
Les laboratoires d’analyses médicales utilisent une centrifugeuse pour séparer les constituants
solides du sang (plaquettes, globules rouges et globules blancs) de son constituant liquide (le plasma) ou encore de purifier des virus.
METHODES DE RESOLUTION DES MELANGES HOMOGENES :
Les constituants d’un mélange homogène peuvent être séparés par plusieurs techniques :
Séparation par rupture de phase :
La rupture de phase se fait soit par augmentation de la concentration de l’analyte (évaporation partielle ou totale du solvant), soit par diminution de sa solubilité (précipitation).
Évaporation du solvant :
C’est un processus par lequel on élimine la partie liquide d’un mélange homogène en le transformant en gaz.
Pour ce faire, on peut laisser le constituant liquide du mélange s’évaporer naturellement à température ambiante, ou on peut accélérer le processus en chauffant le mélange.
L’évaporation totale sert à récupérer la partie solide d’un mélange hétérogène ou encore le soluté d’une solution.
L’évaporation partielle permet de concentrer le soluté d’une solution dans un plus petit volume de solvant.
Précipitation :
On peut diminuer la solubilité du soluté par des moyens physiques ou chimiques.
Son but est de transformer un mélange homogène en un mélange hétérogène facile à séparer par les moyens discutés précédemment.
La précipitation chimique consiste à former une phase hétérogène au sein d’une phase homogène par l’ajout d’un autre soluté qui pourra réagir avec l’analyte pour former une substance solide.
Les moyens physiques consistent en ; la modification de la température, l’addition d’un non solvant ou le relargage.
Modification de la température :
La quantité de soluté pouvant être dissoute dépend de la quantité de solvant et de sa température.
En règle générale, plus haute est la température et plus grand le volume de solvant, plus de soluté peut être dissous.
Une solution qui ne peut plus dissoudre de soluté à une certaine température est dite saturée.
Si l’on refroidit une solution saturée ou si on l’évapore, elle ne sera plus capable de garder le montant originel de soluté et une partie de celui-ci se cristallisera hors de la solution.
Les impuretés cependant resteront dans la solution et ne se retrouveront pas dans les cristaux qui seront purs.
Addition d’un non solvant :
On ajoute un solvant qui diminue la solubilité de l’analyte au sein du mélange.
On récupère donc facilement le solide formé.
Relargage :
C’est une technique qui consiste à séparer une substance en solution de son solvant en introduisant une autre substance plus soluble qui prend sa place.
Lorsqu’une substance est en solution, chaque molécule est entourée par des molécules de solvant qui l’empêchent de se grouper.
Si on introduit dans la solution une substance plus facilement soluble que la première, celle-ci monopolise les molécules du solvant permettant à la première de se séparer du solvant (phénomène de compétition).
Le relargage peut être suivi d’une décantation, une filtration ou d’une distillation.
Parfois deux phases liquides dont l’une est aqueuse ont du mal à se séparer du fait de leurs densités voisines.
On sature la phase aqueuse avec du chlorure de sodium et la séparation est facilitée.
Cette technique est utilisée pour séparer l’ADN ou les protéines (en ajoutant du sulfate d’ammonium).
Séparation par changement d’état :
Distillation simple :
C’est une technique utilisée pour séparer les constituants d’un mélange homogène liquide ou d’un mélange hétérogène comportant au moins une phase liquide.
En utilisant cette technique, on fait appel à la propriété de point d’ébullition.
La distillation ressemble à l’évaporation.
La seule différence est que la vapeur est récoltée par condensation au contact
d’une surface froide.
Elle repose sur deux changements d’état inverses : la vaporisation et la liquéfaction.
Elle nécessite un montage un peu complexe (Voir Figure.7.).
Le mélange homogène liquide est chauffé grâce au chauffe ballon jusqu’à atteindre le point d’ébullition du liquide le plus volatile.
Ce liquide se vaporise donc et monte le long de la colonne puis la vapeur est condensé (distillat) en passant à l’intérieur du réfrigérant grâce à un circuit externe dans lequel circule en permanence de l’eau fraîche.
Le deuxième liquide n’atteint pas sa température d’évaporation et reste sous forme liquide dans le contenant initial (résidu).
En contrôlant la température avec un thermomètre en haut de la colonne, on sélectionne les corps que l’on souhaite récupérer dans la fiole placée à la sortie du réfrigérant.
Le bulbe du thermomètre est disposé tout près du tube à dégagement pour mesurer la température de la vapeur à condenser.
D’habitude on ajoute au ballon de petits morceaux de pierre ponce pour réguler l’ébullition.
Figure.7. Montage de distillation
Distillation fractionnée :
Par simple distillation, on ne peut séparer de façon satisfaisante un mélange d’eau et d’éthanol.
Cela s’explique par la différence relativement faible entre leurs points d’ébullition (100 °C pour l’eau et 78 °C pour l’éthanol).
Quand le mélange est chauffé à 78 °C, l’eau et l’alcool s’évaporent tous deux.
Cependant, la présence de la colonne de fractionnement fournit des surfaces suffisamment froides pour que le composant le moins volatil de la vapeur, l’eau, s’y condense.
Finalement, en théorie, seul le composant le plus volatil, l’alcool, atteint le haut de la colonne et passe dans le réfrigérant.
En réalité, de la vapeur d’eau réussit toujours à accompagner l’alcool.
En recommençant plusieurs fois à partir du distillat, on arrivera cependant à des concentrations assez fortes en alcool.
Sublimation :
Quelques solides passent directement par chauffage à l’état gazeux sans passer par la phase liquide.
Cette sorte de solide peut être séparée d’un mélange en chauffant ce dernier au-delà du point de sublimation et en fournissant une surface relativement froide pour la sublimation inverse (condensation).
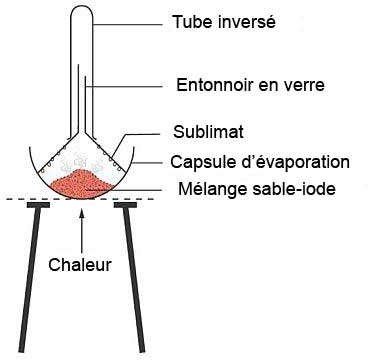
Figure.8. Sublimation
Séparation par transfert de phase :
C’est une méthode de l’analyse immédiate qui consiste à transférer sélectivement un composé ou plusieurs composés du mélange homogène d’une phase à une autre phase (réalisation d’équilibres entre deux phases).
Ce transfert se base sur des propriétés physico- chimiques des composés.
Les deux phases peuvent être deux liquides (extraction par un solvant non miscible, chromatographie de partage liquide-liquide), un liquide et un solide (dissolution fractionnée, chromatographie d’adsorption liquide-solide), un liquide et un gaz (adsorption liquide-gaz, chromatographie phase gazeuse).
Toutes les méthodes chromatographiques vont être entamées en détail en troisième année.
Dissolution fractionnée :
La dissolution fractionnée est utilisée pour séparer un mélange de solides, dans lequel l’un est soluble dans un solvant et l’autre ne l’est pas.
C’est le cas d’un mélange d’hydroxyde de potassium et de carbonate de Calcium.
Comme l’hydroxyde est assez soluble dans l’eau (ce qui n’est pas le cas du carbonate), en ajoutant cette substance au système, l’hydroxyde se dissout.
Par filtration, le carbonate, le composant non dissolvant, est séparé.
Elle consiste à utiliser initialement des liquides à faible pouvoir solvant puis à augmenter progressivement la capacité de dissolution par l’emploi des solvants de plus en plus actifs.
Adsorption :
Principe :
L’adsorption, à ne pas confondre avec l’absorption, est un phénomène physique de surface par lequel des substances (adsorbats) se fixent sur une surface solide (adsorbant) depuis une autre phase (gazeuse, liquide ou solide).
Cette propriété est liée à la structure de l’adsorbant.
Des interactions adsorbant-adsorbats réversibles de faible intensité (Van der Waals, les interactions dipolaires) s’établissent, c’est l’adsorption physique (physisorption) non destructive.
L’adsorption chimique (chimisorption) qui est irréversible met en jeu des forces de plus grande intensité (liaison covalente), et modifie la structure moléculaire du soluté.
La physisorption est utilisée pour les composés organiques alors que la chimisorption est utilisée pour les composés minéraux.
Le phénomène inverse, par lequel les molécules adsorbées sur une surface solide s’en détachent, notamment sous l’action de l’élévation de la température, ou de la baisse de pression, se nomme la désorption.
Une molécule d’adsorbat attirée inégalement par les molécules de deux phases trouvera une position énergétiquement favorable à la surface de l’adsorbant.
Si les conditions énergétiques ou cinétiques permettent à la molécule de pénétrer au sein d’une phase liquide ou solide, il y a absorption.
L’adsorption est favorisée pour :
l’adsorbant, par une faible granulométrie (la taille des grains doit être proche de la taille moléculaire), donc une grande surface spécifique (la surface accessible aux solutés doit être maximale),
l’adsorbat, une masse moléculaire élevée.
L’équilibre adsorption-désorption est influencé par les facteurs de température et de concentration.
La quantité adsorbée augmente quand la température diminue (réaction exothermiques).
Plus la concentration est élevée plus l’adsorption est importante jusqu’à la saturation de l’adsorbant.
L’efficacité se mesure en pourcentage de la masse de produit adsorbé par rapport à la masse de l’adsorbant.
Elle est en générale inférieure à 30 %.
Bien que les phénomènes fondamentaux soient les mêmes, l’adsorption se manifeste différemment sur les solides et sur les liquides ; les méthodes d’étude et les domaines d’application sont différents et justifient des traitements distincts.
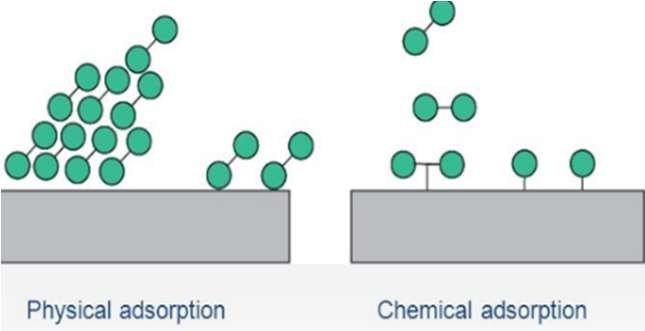
Figure.9. Phénomène d’adsorption
Matériaux adsorbants :
Les principaux matériaux adsorbants que l’on trouve sur le marché sont ; les charbons actifs, les matériaux à base de fibres de carbone, les zéolithes, le gel de silice, alumine active et les adsorbants synthétiques (résines, macromolécules).
Le principal adsorbant utilisé en pratique est le charbon actif, obtenu à partir de matières organiques (bois, tourbe) carbonisées, puis activées.
Le charbon actif peut être obtenu sous forme de poudre ou de grain.
Il peut être régénéré (par désorption).
Isothermes d’adsorption :
L’étude de l’adsorption est basée sur la mesure de la corrélation entre la concentration d’adsorbat dans la phase fluide et la quantité d’adsorbat qui est piégée par la surface à une température donnée : c’est la mesure des isothermes d’adsorption.
De très nombreux modèles ont été créés, certains sont très simples alors que d’autres nécessitent des calculs très complexes.
Ces isothermes d’adsorption/désorption présentent en général trois zones, chaque zone correspondant à un mode de fixation particulier de l’eau sur le produit :
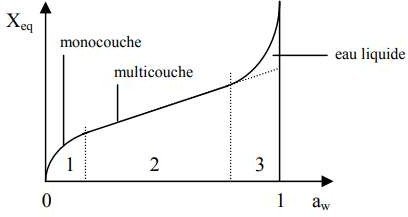
Figure.10. Forme générale des isothermes d’adsorption/désorption
Les isothermes d’adsorption dépendent de la nature du solide (adsorbant) et de l’adsorbat.
On aura ainsi ;
Phase gazeuse :
Pour une température donnée, l’équation caractéristique décrivant le phénomène d’adsorption depuis la phase gazeuse est la quantité d’adsorbat capturée par la surface de l’adsorbant en fonction de la pression d’adsorbat :
𝑓(𝑃 ) = nA , Où nA étant le nombre de moles adsorbées et mS la masse d’adsorbant.
A mS
Six formes caractéristiques ont été définies (voir tableau.3.).
On constate que dans certains
cas les isothermes d’adsorption sont différents des isothermes de désorption, le phénomène d’adsorption n’est donc pas toujours parfaitement réversible.
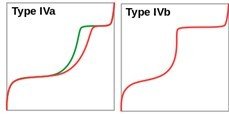
Tableau.3. Classification des isothermes d’adsorption (en rouge) et de désorption (en vert)
Types Formes
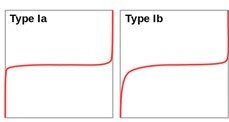
I : adsorbants microporeux (porosité < 2 nm)
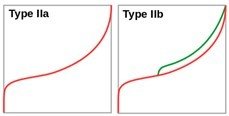
II : des agrégats de grains non poreux (argiles, ciments, pigments)

III : adsorption rare de très faible énergie sur un échantillon non poreux
IV : des matériaux mésoporeux (2 < porosité < 50 nm)
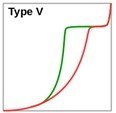
V : similaire au type IV mais de très faible énergie
VI : il indique la formation successive de couches adsorbées (isotherme à marches).
Phase liquide :
Si l’adsorbat est une molécule en solution dans un solvant, l’équation caractéristique devient en fonction de la concentration d’adsorbat (CA) :
𝑛A
𝑓(𝐶 ) =
Il y’a quatre groupes principaux :
A 𝑚S
Type S : adsorption faisant intervenir des interactions adsorbat-adsorbant et adsorbat-adsorbat.
Type L : isotherme de Langmuir.
Type H : forte affinité entre l’adsorbat et l’adsorbant (adsorption importante même à faible concentration).
Type C : isotherme linéaire.
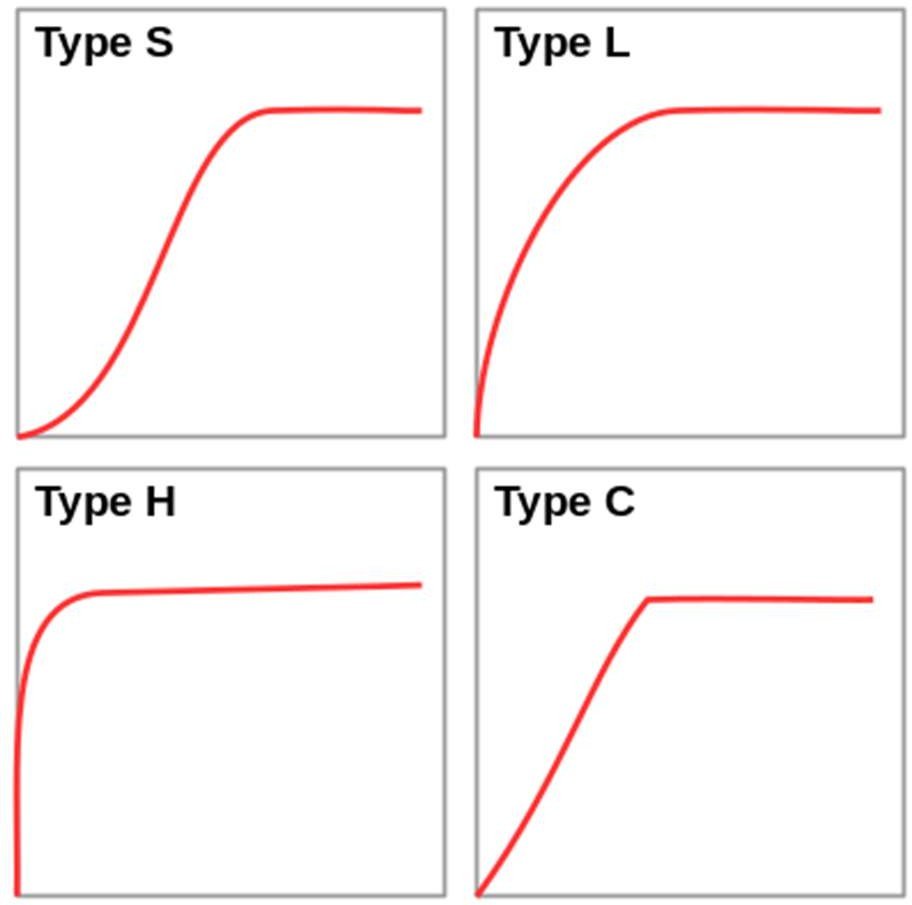
Figure.11. Principaux isothermes d’adsorption liquide
Applications :
Ce phénomène a une très grande importance dans de nombreux processus physiques et chimiques : capture de polluants, séparation de gaz, catalyse, etc.
Il est aussi la base de nombreuses méthodes de caractérisation des solides comme la mesure des surfaces spécifiques ou l’étude de la porosité.
En mécanique industrielle, il joue un rôle fondamental dans les processus de lubrification et les procédés de brasage.
L’adsorption est essentiellement utilisée pour l’épuration des eaux résiduaires et pour dépolluer l’air.
Extraction liquide-liquide :
L’homme utilise des colorants, des parfums, des arômes, et des extraits de produits naturels depuis la haute Antiquité.
Les extractions sont parmi les méthodes les plus utilisées en analyse chimiques.
En effet, Il en existe deux procédés, l’extraction solide-liquide et l’extraction liquide-liquide.
Figure.12. Les différents procédés d’extraction
L’extraction solide-liquide est un phénomène lent qui permet d’extraire une substance présente dans un solide pour la faire passer dans un solvant liquide (exemples ; la macération, l’infusion et la décoction).
L’extraction liquide-liquide est une opération fondamentale de transfert de matière entre deux phases liquides qui permet de séparer un ou plusieurs constituants en fonction de leur différence d’affinité entre les deux phases.
Cette technique permet donc d’extraire une substance dissoute dans un solvant, à l’aide d’un autre solvant, appelé solvant d’extraction ou solvant extraxtif, dans lequel elle est plus soluble.
Tous les liquides ne se mélangent pas nécessairement, ces liquides sont dits non-miscibles et forme deux phases distinctes superposées en fonction de leur densité, contrairement aux liquides miscibles qui se mélangent bien pour former une solution homogène.
En fonction de cette miscibilité, on distingue l’extraction par solvants miscibles et celle par solvants non miscibles.
Figure.13. Miscibilité de quelques solvants
Extraction par un solvant miscible :
Ce sont des techniques de purification de solutions par effet de diffusion à travers une membrane.
On distingue différents types de membranes.
Les membranes hémiperméables qui ne laissent passer que le solvant.
Les membranes dialysantes ayant des pores de diamètre identiques et connus qui laissent passer le solvant et les solutés en dessous d’une certaine taille.
Osmose et osmose inverse :
L’osmose est un procédé de séparation en phase liquide par transfert de solvant à travers une membrane hémiperméable sous l’effet d’un gradient de concentration.
Si on considère un système à deux compartiments séparés par une membrane semi-sélective et contenant deux solutions de concentrations différentes, l’osmose se traduit par un flux de solvant résultant d’un effet de diffusion de la solution diluée (hypotonique) vers la solution concentrée (hypertonique).
Ce phénomène s’arrête lorsque les deux liquides ont atteint la même concentration (milieux isotoniques), ce qui revient à équilibrer les pressions osmotiques.
Un même phénomène devrait se produire en sens inverse pour le soluté, mais la membrane n’est pas perméable aux molécules de soluté.
Si on applique une pression sur la solution concentrée, la quantité de solvant transférée par osmose va diminuer.
Avec une pression suffisamment forte, le flux de solvant va même s’annuler : cette pression est nommée la pression osmotique Π.
Si on dépasse la valeur de la pression osmotique, on observe un flux d’eau dirigé en sens inverse du flux osmotique : c’est le phénomène d’osmose inverse.
Comme son nom le suggère, les échanges de solvant se font par perméation sous l’effet d’un gradient de pression depuis le milieu hypertonique vers le milieu hypotonique, ce qui revient à concentrer davantage le milieu le plus concentré.
Le liquide est refoulé au travers de la membrane, laissant les solides dissous derrière.
L’osmose inverse utilise des membranes denses sans porosité qui laissent passer le solvant et arrêtent les ions.
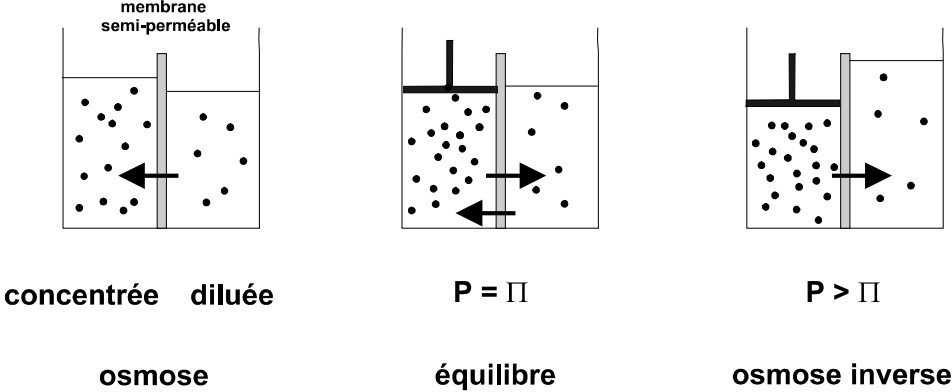
Figure.14. Phénomène d’osmose et d’osmose inverse
La pression osmotique en pascals est donnée par la relation suivante : Π= iCRT, où i est le nombre d’espèces d’ions constituant le soluté, C la concentration molaire du soluté, T la température (K) et R la constante des gaz parfaits (8,31 J.mol-1.K-1).
Cette relation est valable pour des solutions diluées.
Une partie de la solution à traiter (débit Q0) se divise au niveau de la membrane en deux parties de concentrations différentes.Une partie (débit Qp) passe à travers la membrane (perméat) et une partie retenue (concentrat ou rétentat).
La fraction de débit qui traverse la membrane est le taux de conversion Y défini par :
𝑌 =
𝑄P
𝑄0
La sélectivité d’une membrane est définie par le taux de rejet R (ou taux de rétention):
𝑅 = C0–CP = 1- CP
C0 C0
Où C0 est la concentration de l’espèce à retenir dans la solution et Cp est la concentration de la même espèce dans le perméat.
L’osmose inverse est principalement utilisée dans le dessalement de l’eau de mer et des eaux saumâtres, la production de l’eau ultrapure (industries électronique, pharmaceutique …) et dans l’extraction de protéines du lactosérum dans l’industrie laitière.
Dialyse :
Par effet de diffusion, les petites molécules traverseront la membrane dialysante, tandis que les grosses molécules seront retenues.
Le produit d’une dialyse (solution de recueil des petites molécules) s’appelle un dialysat.
La principale application de la dialyse dans le domaine médical concerne les personnes souffrant d’une insuffisance rénale (élimination des déchets).
Electrodialyse :
C’est un processus membranaire, au cours duquel les ions sont transportés à travers une membrane semi-perméable, sous l’influence d’un potentiel électrique.
Les membranes sont sélectives pour les cations ou les anions, rejettent les ions de même charge.
Les membranes sélectives pour les cations sont constituées de polystyrène sulfoné, tandis que les membranes sélectives pour les anions sont constituées de polystyrène avec de l’ammoniac quaternaire.
Les particules qui ne portent pas de charge électrique ne sont pas éliminées.
Cette technique peut être appliquée pour éliminer les ions de l’eau.
Extraction par un solvant non-miscible :
Principe :
Elle permet de transférer sélectivement, sous l’effet de la solubilité, un ou plusieurs soluté(s) d’une phase liquide à une autre phase liquide non-miscible à la première.
Elle est d’autant plus efficace que ce soluté est plus soluble dans le solvant extractif que dans son solvant original.
Par conséquent, on choisit lorsque cela est possible, un solvant extractif dans lequel le soluté est très soluble.
Cette technique requiert une succession de plusieurs étapes.
La première étape est la mise en contact intime des deux solvants non miscibles durant un temps suffisant.
La seconde étape consiste à obtenir l’équilibre de partage du soluté établit entre les deux solvants.
Cet équilibre
est régi par les lois de la diffusion et de la solubilité.
La dernière étape est la séparation des phases en fonction de leur densité (d) par décantation.
La densité est une grandeur sans unité qui reflète la capacité d’un solvant à flotter ou à couler dans l’eau.
C’est donc le rapport des masses volumiques des deux liquides.
Cela permettra de savoir si le solvant extractif se retrouve au-dessus ou en-dessous de la phase à extraire.
On en déduira que, dans un mélange de liquides non miscibles :
-Si d<1, le solvant se trouvera au-dessus de l’eau.
-Si d>1, le solvant se trouvera au-dessous de l’eau.
Même si le principe de l’extraction liquide-liquide parait simple, sa mise en place est assez complexe.
Il faut bien choisir le système liquide-liquide, le procédé et enfin l’appareil qui donneront les meilleures performances.
Choix du solvant extractif :
Ce choix repose sur différents paramètres ;
Son état physique ; il doit être liquide à la température et la pression de l’extraction.
Sa non miscibilité avec à la phase à extraire.
Sa capacité à dissoudre facilement l’espèce à extraire.
Facilement éliminés après extraction, ayant un point d’ébullition bas et le plus éloigné possible de celui l’espèce à extraire.
Son inertie chimique vis-à-vis de la solution à extraire.
Sa toxicité, on choisit le solvant le peu toxique que possible.
Son cout, on choisit lorsque cela est possible le solvant le moins cher possible.
Etude de l’équilibre de partage :
La répartition inégale du soluté entre les deux phases liquides non-miscibles repose sur sa solubilité dans ces deux liquides.
La solubilité d’un corps pur dans un solvant est fonction de sa polarité.
Deux composés de polarité proche sont solubles entre eux.
On utilise habituellement une phase aqueuse et une phase organique.
Les produits organiques neutres sont moins polaires et donc beaucoup plus solubles dans les solvants organiques.
Les sels inorganiques et les composés organiques chargés sont plus solubles dans l’eau que dans les solvants organiques, d’où l’intérêt de connaître le pKa des produits et le pH des solutions.
Lors de la mise en présence d’un soluté et de deux liquides non miscibles, il s’établit un équilibre caractérisé par une constante thermodynamique appelée le coefficient de partage (Kp).
Ce dernier est le rapport des concentrations du soluté dans chacune des deux phases.
Le graphe représentant l’équilibre de partage CB= f(CA) (avec CA et CB sont les concentrations du soluté respectivement dans les phases A et B à l’équilibre) est soit ;
-Une droite dans le cas d’une distribution régulière.
Le rapport CB/CA est constant et représente le coefficient de partage (CB = Kp CA).
-Une courbe dans le cas d’une distribution irrégulière, cela signifie que le soluté à extraire n’existe pas sous la même forme dans les deux phases A et B.
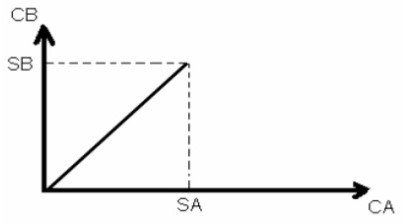

Figure.15. Graphes de distribution régulière (à gauche), irrégulière (à droite)
Soit un produit S qu’on veut extraire d’une phase A à l’aide d’un solvant B. m0 est la masse initiale du composé S dans la phase A, mA la masse restante du composé S dans la phase A après une extraction, mB la masse du composé extrait par le solvant B.
On a ;
Kp = [S]B
| SA | S B | |
| Avant agitation : | m0 | 0 |
| A l’équilibre : | mA, [S]A | mB, [S]B |
[S]A
Kp = nB/VB= mB/VB
avec m0= mA + mB
nA/VA
mA/VA
Kp = m0–mA/VB
mA/VA
Donc mA= m0 VA
Kp(VB+VA)
L’extraction sera d’autant plus efficace que le coefficient de partage est grand.
On ne connaît habituellement pas le coefficient de partage du produit.
On utilise souvent le même volume de solvant d’extraction que celui de la phase à extraire ou un volume deux fois supérieur.
On effectue normalement 2-3 extractions de la phase aqueuse.
On peut vérifier par CCM qu’il ne reste plus de produit dans la phase aqueuse.
On peut modifier le coefficient de partage du produit par relargage en ajoutant un peu d’une solution aqueuse saturée en NaCl.
Coefficient de partage relatif ou corrigé α :
Le coefficient de partage relatif est le rapport des quantités. 𝑄A et 𝑄B sont les quantités de soluté S respectivement dans les phases A et B à l’équilibre.
α = QB = [S]BVB = Kp VB
Taux de distribution D :
QA [S]AVA VA
Lorsque S se trouve sous plusieurs formes (moléculaire et ionisée) dans une et/ou l’autre des phases, il faudra tenir compte de la somme des concentrations de chaque forme et il convient de parler de la constante de distribution D définie par l’équation suivante :
[S]B(moléculaire)+ [S]B(ionigue)
D = ΣC /ΣC =
B A [S]A + [S]A
(moléculaire) (ionigue)
Rendement d’extraction R :
L’efficacité d’une extraction est évaluée par son rendement.
On cherche souvent à récupérer la plus grande quantité possible de soluté S à l’aide d’un volume donné de solvant extractif.
Le rendement d’extraction est la quantité de soluté S extrait par le solvant B (QB) à partir de la quantité initiale présente dans le solvant A (QA0).
Plus QB sera proche de QA0, meilleur sera le rendement d’extraction.
R= ZQB
QA0
Le rendement d’extraction dépend du solvant et du soluté et par conséquent de la constante
de distribution, du volume de solvant d’extraction et du nombre d’opérations effectuées.
Notion d’étage théorique :
Un étage théorique est l’opération successive agitation et décantation.
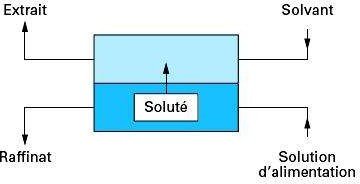
Figure.16. Schéma d’une opération d’extraction à un seul étage
Les principaux procédés d’extraction par un solvant non miscible :
Il existe différents processus d’extraction liquide par un solvant non miscible ; extraction simple, répétée et à contre-courant.
Ces techniques permettent la récupération simultanée de plusieurs molécules.
Cependant ces techniques ne sont pas strictement spécifiques d’une molécule car le solvant employé peut extraire de la matrice toutes les molécules ayant des propriétés physico-chimiques proches.
Le choix du type d’extraction dépend des applications.
Extraction simple :
Principe :
Elle consiste à extraire en une seule étape le maximum de soluté initialement présent dans la solution à extraire par le solvant extractif.
Le solvant extractif n’est donc mis qu’une seule fois en contact de la solution à extraire (étage théorique unique).
Le solvant extractif sera choisi en fonction de son pouvoir dissolvant vis-à-vis de soluté.
En pratique, le solvant A et le solvant B sont mis en contact dans une ampoule à décanter, puis une agitation est pratiquée pendant un temps nécessaire à l’établissement d’équilibre.
Cependant l’agitation ne doit pas être trop brutale et doit être réalisée de manière à éviter les émulsions qui sont ensuite difficiles voire impossibles à casser.
La formation d’émulsions est un phénomène fréquent lors d’extraction de matrice biologique, rendant la méthode d’extraction non reproductible.
Toutefois les températures de congélation des solvants d’extraction étant plus basse que celles des matrices biologiques, l’extrait peut être congelé afin de faciliter la récupération de la phase organique.
La séparation des deux phases s’effectue par une décantation.
L’extrait est récupéré puis concentré par évaporation partielle ou complète.
Etude quantitative :
| Phase A à extraire | Phase B extractive | |
| 1 étage théorique | SA | S B |
| Avant agitation : | CA0 ou QA0 | CB0 ou QB0=0 |
| A l’équilibre : | CA1 ou QA1 | CB1 ou QB1 |
Cas d’une distribution régulière :
On a ; VA est le volume de la phase à extraire et VB est le volume de la phase extractive.
QA0 = QA1 + QB1
Kp = CB1
CA1
α = QB1 = CBVB = Kp VB
QA1
CAVA VA
R= QB1 = CB1VB
QA0 CA1VA+ CB1VB
R = 1 − 1
[1+𝐾𝑝 VB]
VA
Cas d’une distribution irrégulière :
= α 1+α
= 1- 1
1+α
On utilise la méthode graphique pour la détermination des concentrations CA1 et CB1.
On a ; QB1= QA0 – QA1= VA (CA0 – CA1) = CB1 VB CB1 = VA (CA0 – CA1)
VB
A partir de la courbe : CB =f (CA), pour CA1=0 CB = VA CA0
VB
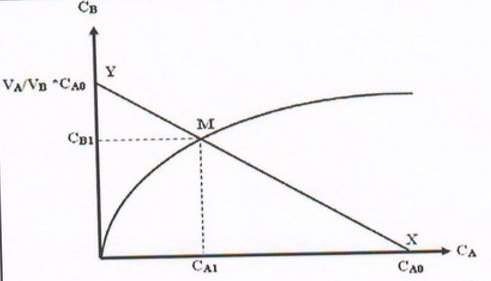
Figure.17. Détermination graphique des concentrations CA1 et CB1
Cette technique peut être mise à profit afin d’extraire, en une seule étape, un groupe de molécules de la même famille chimique (les benzodiazépines, les lipides ou les pesticides).
Extraction répétée :
Principe :
La solution à extraire est mise en contact plusieurs fois avec le solvant extractif (n opérations, n étages théoriques).
Toute nouvelle portion utilisée de celui-ci est vierge du soluté à extraire.
Les extractions multiples sont employées dans deux cas ;
Le rendement d’extraction simple n’est pas satisfaisant.
Il peut être nécessaire de répéter l’opération d’extraction.
On parle alors d’extraction par épuisement.
Les différents extraits obtenus sont par la suite mélangés puis concentrés pour isoler le soluté.
L’inconvénient majeur de cette méthode est l’utilisation de gros volume de solvant, ainsi qu’une augmentation du temps de traitement de l’échantillon.
L’extrait n’est pas suffisamment purifié, la réextraction de ce premier est nécessaire afin d’en éliminer les substances interférentes.
Le soluté est en général resolubilisé dans une phase aqueuse.
Cette procédure est appelée back-extraction.
Cependant, il faut garder en mémoire que l’ajout d’étapes purifie certes l’échantillon, mais augmente le temps de prétraitement de l’échantillon et le risque de perte du soluté.
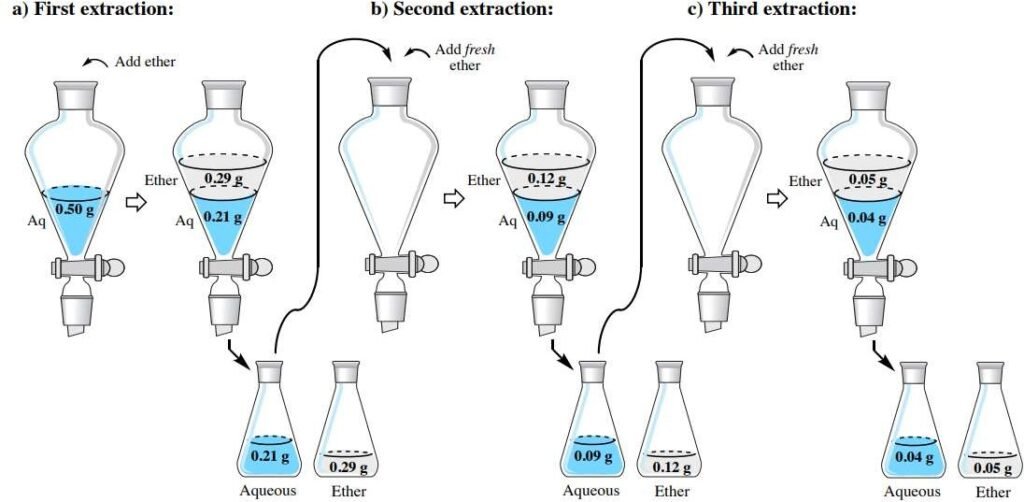
Figure.18. Exemple schématique d’extraction répétée
Etude quantitative :
| Phase A à extraire | Phase B extractive | |
| 1 èr étage | SA | S B |
| Avant agitation : | QA0 | QB0=0 |
| A l’équilibre : | QA1 | QB1 |
| 2 ème étage | SA | S B |
| Avant agitation : | QA1 | 0 |
| A l’équilibre : | QA2 | QB2 |
| n ème étage | SA | S B |
| Avant agitation : | QA(n-1) | 0 |
| A l’équilibre : | QAn | QBn |
Cas d’une distribution régulière :
Le rendement du processus d’extraction répétée à n étages théoriques sera calculer ainsi ; On a ; VB1 ≠ VB2≠ VB3≠ VB4≠ VBn
QA0 = QAn + ∑QB = QAn + [QB1+ QB2 + + QB (n-1) + QBn]
Kp = CB1 = CB2 = CBn
CA1 CA2 CAn
α1 = QB1 , α2 = QB2 , ………,αn= QBn
QA1
QA2
QAn
QA0 (1– 1 )
R= ∑QB = (1+α1)(1+α2)(1+α3) (1+αn)
QA0 QA0
R = 1- 1
(1+α1)(1+α2)(1+α3) (1+αn)
Si on utilise le même volume de solvant extractif VB dans chaque étage théorique, la formule du rendement devient ;
α = QB1 = QB2 = QBn
QA1 QA2 QAn
R = 1 – 1
[1+α]n
= 1 – 1
VB n
[1+𝐾𝑝 ]
VA
Pour un même volume de solvant organique VB, le rendement d’extraction est meilleur lorsqu’une extraction multiple est réalisée par rapport à une extraction simple.
Figure.19. Schéma d’une extraction répétée à 3 étages
Extraction à contre-courant :
La solution à extraire circule en sens inverse du solvant extractif dans une tour d’extraction.
Il s’ensuit que la partie la moins riche en soluté de la solution à extraire est en contact avec le solvant le plus pur ainsi que les parties les plus riches des deux solutions, mais à l’autre extrémité de la tour.
Cette méthode sera encore plus détaillée en troisième année.
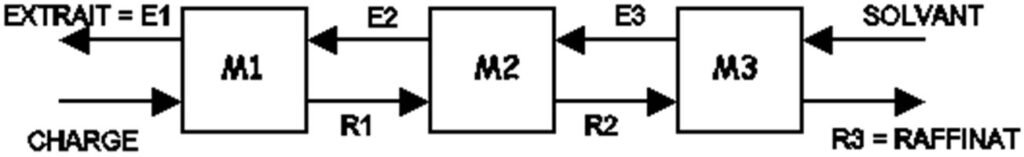
Figure.20. Schéma d’une extraction à contre-courant à 3 étages
Avantages et inconvénients de l’extraction liquide-liquide :
Cette méthode de séparation présente de nombreux avantages parmi lesquels :
Le coût : ce sont des techniques qui ne demandent pas d’investissement de gros matériel ou de réactifs.
La concentration des échantillons : l’utilisation de solvants organiques volatiles permet la concentration du soluté par évaporation du solvant.
La purification : l’utilisation d’un solvant organique judicieusement choisi permet de solubiliser la substance d’intérêt et de laisser dans la matrice les molécules interférentes.
La possibilité de travailler sur des matrices très variées (sang total laqué post mortem, viscères ou cheveux) qui ne sont pas toujours compatibles avec l’extraction en phase solide.
La possibilité d’extraire une gamme très étendue de molécules qui couvre une multitude d’applications (industrie pharmaceutique, nucléaire, pétrochimique…etc.).
L’extraction liquide-liquide présente aussi quelques inconvénients ;
La consommation de volumes importants de solvants, surtout lorsqu’il s’agit d’extractions multiples : cet inconvénient peut être minimisé par la diminution de la prise d’essai.
La toxicité des solvants.
Difficultés d’extraire les molécules très polaires de part les caractéristiques chimiques des solvants organiques (apolaire et aprotiques).
Ce sont des techniques manuelles, consommatrices de temps et de personnel.
Applications de l’extraction liquide-liquide :
Cette technique efficace et peu coûteuse a fait l’objet de plusieurs applications importantes tant du point de vue quantitatif (pétrochimie) que qualitatif (industries alimentaires et pharmaceutique, récupération des polluants dans des effluents d’usine).
Elle est utilisée dans la fabrication de plusieurs composés chimiques dont les acides minéraux (acide phosphorique très pur), les acides organiques (acide carboxylique, acide gallique…etc), le brome, le nitrate de potassium et les acides nitrofluorydriques.
L’extraction est le plus souvent utilisée lorsque les constituants à traiter sont thermosensibles (antibiotiques) ou que les points d’ébullition sont proches (séparations d’aromatiques).
Ce procédé́ permet la concentration et la purification de ;
Principes actifs pharmaceutiques d’origine synthétique ou naturelle.
La découverte de nouveaux médicaments peut passer par l’étude des substances naturelles et si une molécule se trouve être performante dans un domaine précis, elle pourra faire l’objet d’une commercialisation sous forme de médicament.
Les plantes sont mélangées à un solvant capable de traverser la cellule pour en récupérer les substances emprisonnés à l’intérieure.
Ces substances sont ensuite isolées et purifiées par extraction liquide- liquide.
Plusieurs autres molécules (pesticides, stéroïdes, lipides, vitamines, métaux. . .etc).
Les échantillons (biochimiques, pharmacologiques ou toxicologiques) préalablement à une analyse par méthode chromatographique.
C’est le cas notamment l’extraction de stéroïdes urinaires et des antidépresseurs sérotoninergiques.