

Modélisation et paramètres pharmacocinétiques
Définition :
La pharmacocinétique étudie le devenir des médicaments et de leurs métabolites en fonction du temps à l’intérieur d’un organisme vivant et entier.
Ce devenir résulte de divers processus : absorption, distribution, métabolisme et élimination.
L’étude pharmacocinétique d’une drogue se fait à partir des prélèvements des liquides biologiques effectués à différents temps : c’est la pharmacocinétique quantitative.
Elle repose sur la détermination expérimentale des quantités ou des concentrations de médicaments (ou de métabolites) présents dans le sang ou en général les liquides biologiques.
A partir de ces mesures, et de l’évolution de ces concentrations au cours du temps que vont être calculés les paramètres pharmacocinétique.
Intérêt de l’étude de la pharmacocinétique quantitative :
Etablissement du schéma posologique.
Suivi (monitoring) thérapeutique : adaptation des posologies.
Évaluation de la biodisponibilité et la bioéquivalence (générique/ référence).
Paramètres pharmacocinétiques :
Phase d’absorption :
Coefficient de résorption (facteur f) :
La fraction ou le % de la dose administrée ayant été absorbée.
Coefficient d’extraction E :
Fraction du médicament extraite par un organe à chaque passage et soustraite de la circulation
générale.
Q : dose du médicament
E CL Q

Constante de vitesse d’absorption Ka :
Constante de vitesse du processus total de transfert du médicament, de l’extérieur à l’intérieur de l’organisme à travers toute membrane biologique.
Biodisponibilité (facteur F) :
C’est la fraction de la dose du médicament administré qui atteint la circulation générale suite à une administration extravasculaire et la vitesse avec laquelle se produit ce phénomène.
Exprimée par le facteur F: 0-100% (ou entre 0 et 1).
On distingue :
Biodisponibilité absolue : permet d’évaluer l’intérêt d’une voie d’administration par rapport à la voie de référence (I.V).
F (voie IV) = 100% (absorption totale)
F = f x F’
f: coef de résorption
F’ : fraction du PA échappant aux premiers passages
Biodisponibilité relative : est utilisée pour comparer entre elles les performances des formes galéniques identiques ou différentes.
C’est une comparaison entre:
Deux formes galéniques différentes mais pour la même voie d’administration, exemple : comprimé vs sirop.
La même forme galénique fabriquée par deux laboratoires différents (générique vs référence).
Surface sous la courbe (AUC ou SSC):
Surface comprise entre les axes des abscisses et la courbe des concentrations plasmatiques en fonction du temps.
Elle reflète la quantité du médicament ayant atteint la circulation générale.
La surface sous la courbe est soit limitée à un temps déterminé soit extrapolée jusqu’au temps infini.
Phase de distribution :
Constante de vitesse de distribution Kd:
Constante de vitesse du passage du médicament du compartiment central vers les compartiments périphériques.
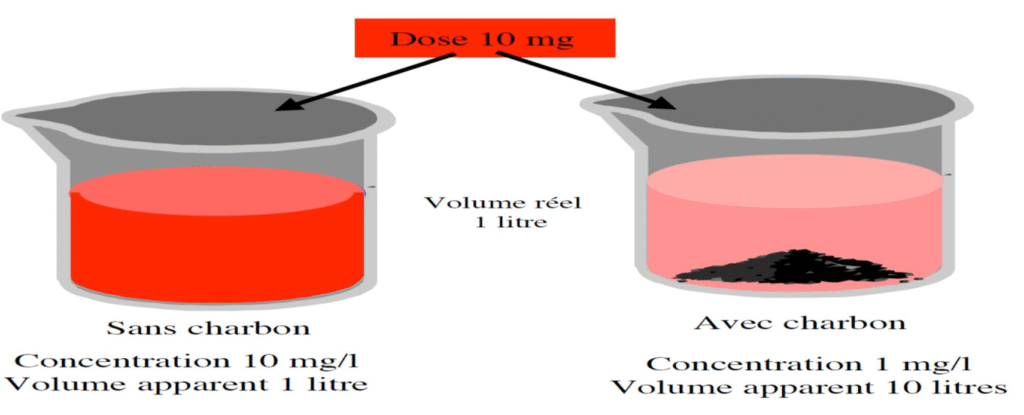
Volume apparent de distribution Vd:
Il est défini à l’état d’équilibre comme étant le volume hypothétique (fictif) de liquide biologique qu’occuperait le médicament s’il avait partout la même concentration que dans le plasma.
C’est une mesure directe de l’importance de la diffusion des médicaments dans l’organisme.
Cependant ce volume ne correspond pas sauf exception ; au volume réel de l’organisme.
Dans certains cas ; le volume de distribution peut être supérieur au volume corporel total.
| Compartiments liquidiens | Volume (L) |
| Plasma | 3 |
| Sang | 5 |
| Lymphe | 10 |
| Eau intracellulaire | 27 |
| Eau extracellulaire | 15 |
| Eau corporelle totale | 42 |
C’est un Facteur de proportionnalité entre la quantité de médicament présent dans l’organisme au temps « t » et la concentration de ce médicament.
C = Q / Vd d’où Vd = Q / C
L’intérêt:
Moyen de quantification de la distribution tissulaire : traduit l’intensité de diffusion du médicament dans l’organisme.

| Compartiments liquidiens | Volume (L) | Exemple |
| Sang | 5 | Poids moléculaire élevé :Héparine ; InsulineForte fixation aux protéines plasmatiques :Phénylbutazone ; Warfarine ; Aspirine |
| Eau extracellulaire | 15 | Théophylline ; Aténolol ; Pénicilline |
| Eau corporelle totale | 42 | Ethanol ; Paracétamol ; Indométacine |
Phase d’élimination :
Constante de vitesse d’élimination Ke :
Constante de vitesse du (des) processus d’élimination du médicament dans l’organisme.
Temps de demi- vie d’élimination T1/2 :
Temps au bout duquel la quantité du médicament est réduite de moitié à la suite d’un processus d’élimination.
L’intérêt :
Indicateur de la durée de séjour du médicament: Au bout de 7 t1/2 environ 99% du médicament est éliminé.
Établissement du rythme posologique (la dose, le nombre et le rythme d’administation)
Estimation du temps d’atteinte du plateau d’équilibre
Clairance Cl. :
C’est le volume de plasma totalement épuré du médicament par unité de temps.

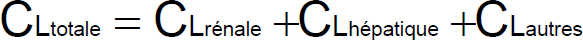
Et: CL =Ke x Vd
Méthodes d’étude de la pharmacocinétique:
Les méthodes morphologiques :
Ces méthodes permettent de situer une substance à un moment donné.
Elles font appel à l’utilisation de molécules marquées par un isotope radioactif.
On emploie de petits animaux, habituellement des souris.
Au moment désiré après l’injection du produit, l’animal est sacrifié par congélation dans l’azote liquide ce qui permet l’interruption immédiate de tous les processus métaboliques.
On peut alors obtenir des images macroscopiques ou microscopiques de la distribution du médicament.
Ces méthodes sont essentiellement qualitatives.
Elles permettent de situer la substance mais ne donnent qu’une idée grossière des quantités contenues dans chaque organe.
Les méthodes physicochimiques :
Elles permettent d’une part l’isolement et l’identification des métabolites, d’autre part le dosage du médicament (et de ses métabolites).
Les prélèvements portent sur le plasma, l’urine, la bile ou encore, chez l’animal, sur les tissus qui sont broyés et homogénéisés.
La plupart des médicaments sont dosables par les méthodes physicochimiques classiques (divers types de chromatographie, spectrophotométrie de masse, spectrofluorimétrie,).
Pour un certain nombre de substances, la radio-immunologie et surtout l’immuno-enzymologie donnent des résultats rapides et précis compatibles avec les besoins cliniques.
Méthodes mathématiques : l’analyse compartimentale
L’analyse compartimentale permet d’établir des « modèles » mathématiques à partir des courbes de concentrations du médicament dans le sang (les urines et les tissus éventuellement) en fonction du temps.
Les modèles doivent permettre de retrouver les résultats expérimentaux
à partir d’expressions mathématiques.
Ils constituent des représentations possibles de la réalité
; on adopte le plus simple compatible avec l’expérience.
Cette méthode repose sur l’idée que le médicament se situe dans un ou plusieurs compartiments, espaces virtuels dans lesquels il est instantanément réparti de manière homogène.
Il s’échange avec les autres compartiments ou s’élimine d’une manière identique (au point de vue cinétique) en tout point de ce compartiment.
Les compartiments n’ont pas forcément de réalité anatomique précise : ce sont des espaces de diffusion virtuels.
On peut calculer le volume du compartiment
; il est le plus souvent fictif.
Modélisation en pharmacocinétique:
Définition d’un modèle:
Les divers aspects du devenir du médicament peuvent être décrits à l’aide d’équations mathématiques.
L’ensemble de ces équations constitue le modèle pharmacocinétique.
Donc le modèle pharmacocinétique est « une représentation mathématique du comportement d’un médicament dans l’organisme ».
Définition du compartiment en pharmacocinétique :
Le modèle en pharmacocinétique fait appel à la notion théorique de compartiment.
C’est un ensemble de molécules ayant la probabilité d’être affectées au même moment par le même événement.
C’est une quantité de tissus ou de liquide théoriques.
On distingue 2 Types de compartiments:
Compartiment central: sang et tissus richement vascularisés (cœur, poumon, foie, rein, cerveau)
Compartiments périphériques: constitués de tissus moins irrigués.
A leur niveau la cinétique du médicament sera différente mais les concentrations présentes dans ces compartiments seront en équilibre avec les concentrations dans le compartiment central.
Ils se divisent en :
Compartiments superficiels: les muscles
Compartiments profonds: les os
Cette subdivision est basée sur le temps nécessaire à l’obtention de l’état d’équilibre entre le compartiment central et les compartiments périphériques.
Modèle pharmacocinétique linéaire ou d’ordre un (1) :
Lorsque la vitesse de transfert du médicament est directement proportionnelle à la quantité, nous sommes donc en présence d’un processus d’ordre premier ; dans ce cas-là le modèle pharmacocinétique est linéaire.
Ceci se traduit par l’équation ;
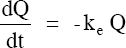

La conséquence d’une cinétique linéaire : L’évolution des concentrations médicamenteuses est prévisible quel que soit le schéma posologique.
Et les paramètres pharmacocinétiques restent constants quel que soit la dose.
Modèle pharmacocinétique non linéaire et ordre de transfert zéro :
La quantité traversant une membrane par unité de temps est constante et indépendante de la quantité de médicament c’est le : processus d’ordre 0 (cinétique non linéaire).
-K0
Modèle monocompartimental:
L’organisme se comporte comme un ensemble homogène.
Administration à dose unique par voie I.V:

C C 0 e
k e t
C
t
Ce modèle ne comporte qu’une seule constante de vitesse qui est celle de l’élimination Ke.
La vitesse de variation de la concentration du médicament par unité de temps est égale au produit de la quantité du médicament par la constante de vitesse de 1er ordre (ke).
Le signe négatif indique que cette concentration diminue dans le temps.
Le logarithme de la concentration varie linéairement avec le temps :
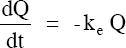
Le volume du compartiment étant constant
dC = – ke C dt
Par intégration ; l’équation devient : C = C0 e -ket
ln C = ln C0 – ke t
t = temps
C = concentration du médicament à l’instant t
C0 = concentration du médicament à l’instant t = 0 Ke = constante de vitesse d’élimination
La représentation graphique en ordonnées semi-logarithmiques permet de linéariser la courbe.
t
logC
Elimination
L’équation (log C = log C0 – ke t) est de la forme :
Y = a X + b (Y = log C ; a : pente de la droite et b= log C0)
Détermination des paramètres pharmacocinétiques :
Calcul de Ke
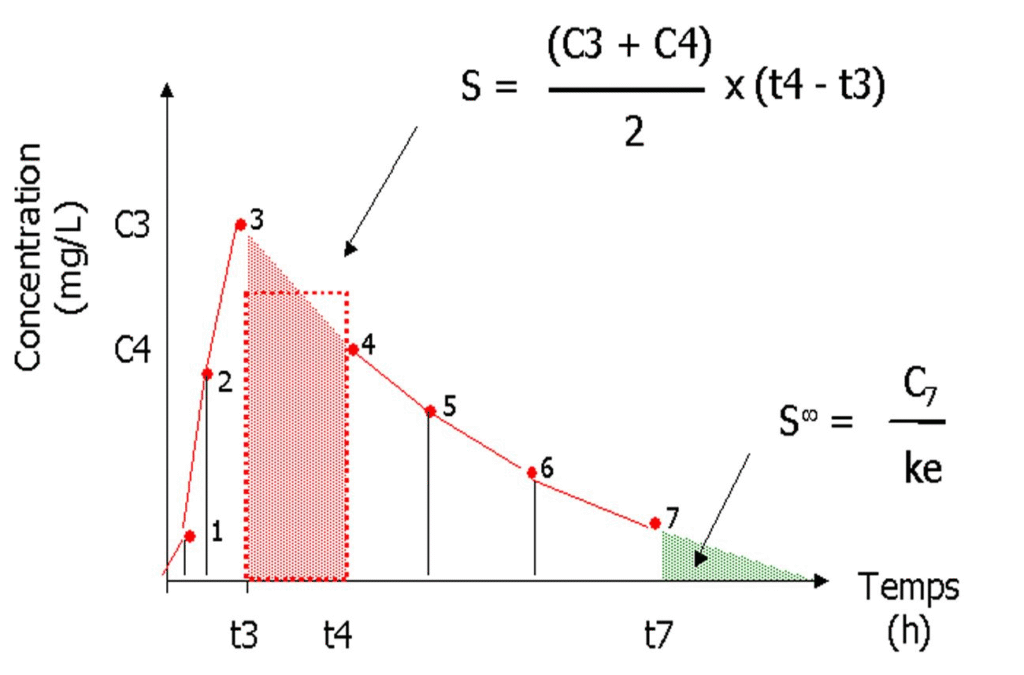
Calcul de la surface sous la courbe :
Deux méthodes de calcul sont possibles :
c1.La méthode graphique dite des trapèzes qui consiste à découper la courbe expérimentale en autant de trapèzes qu’il existe de points expérimentaux :
Surface du trapèze = (C1 + C2) x (t2 – t1)
2
L’addition des trapèzes permet d’obtenir la Surface sous la courbe des points expérimentaux.
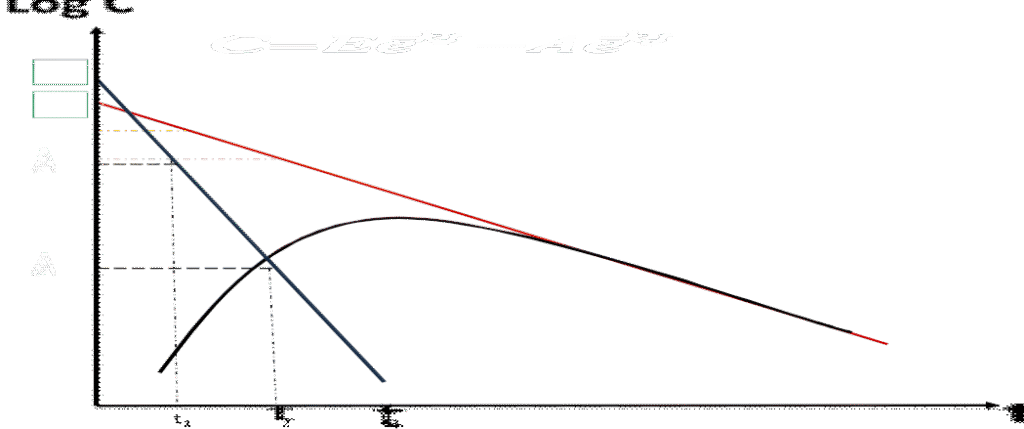
La phase d’élimination est extrapolée (prolongée) jusqu’à l’axe des ordonnée ; l’ordonnée à l’origine correspond au coefficient E.
On établit pour chaque temps de prélèvement de la première phase, la différence entre la concentration plasmatique expérimentale (de la partie ascendante) et la concentration plasmatique théorique obtenue sur la droite d’élimination extrapolée.
La différence obtenue pour chaque point est reportée sur le graphe semi-log.
On obtient une droite qui représente le phénomène d’absorption.
L’intersection de cette droite avec l’axe des ordonnée donne le coefficient A.
Modèle ouvert à deux compartiment
voie orale
Distribution + Élimination
Absorption + Distribution + Élimination
t
Élimination
a phase ascendante correspond au processus d’absorption ; de distribution et d’élimination (mais la vitesse d’absorption est plus importante que celle de distribution et d’élimination).
Donc il faut procéder au lissage de la courbe pour avoir une droite qui représente uniquement le processus d’absorption ; et c’est à partir de cette droite que vont être calculé les paramètres pharmacocinétiques d’absorption (A, Ka).
La première partie descendante correspond au processus de distribution et d’élimination.
Il faut procéder au lissage de la courbe pour avoir une droite qui représente uniquement le processus de distribution ; et c’est à partir de cette droite que vont être calculé les paramètres pharmacocinétiques de distribution (D, Kd).
Détermination des paramètres pharmacocinétiques :
Calcul des constantes de vitesse : Ka ; Kd ; Ke
On procède au lissage de la courbe (non traité dans ce cours)
Calcul de T1/2 :
Le t1/2 est calculé de la même façon que la voie IV.
Calcul de la surface sous la courbe :