
Étude Théorique de la Chromatographie : Tout Comprendre en Pharmacie Analytique
La chromatographie est au cœur de l’analyse pharmaceutique moderne. Pourtant, pour maîtriser cette technique, il ne suffit pas de connaître les protocoles de laboratoire. Comprendre les théories qui la gouvernent — notamment la théorie des plateaux et la théorie cinétique — est indispensable pour optimiser une séparation, améliorer l’efficacité d’une colonne et interpréter correctement un chromatogramme.
Cet article, basé sur le cours du Dr A. Amziane (Université d’Alger I, Faculté de Pharmacie, Janvier 2023), vous guide pas à pas à travers les deux grands modèles théoriques de la chromatographie, avec des explications claires et des exemples concrets.
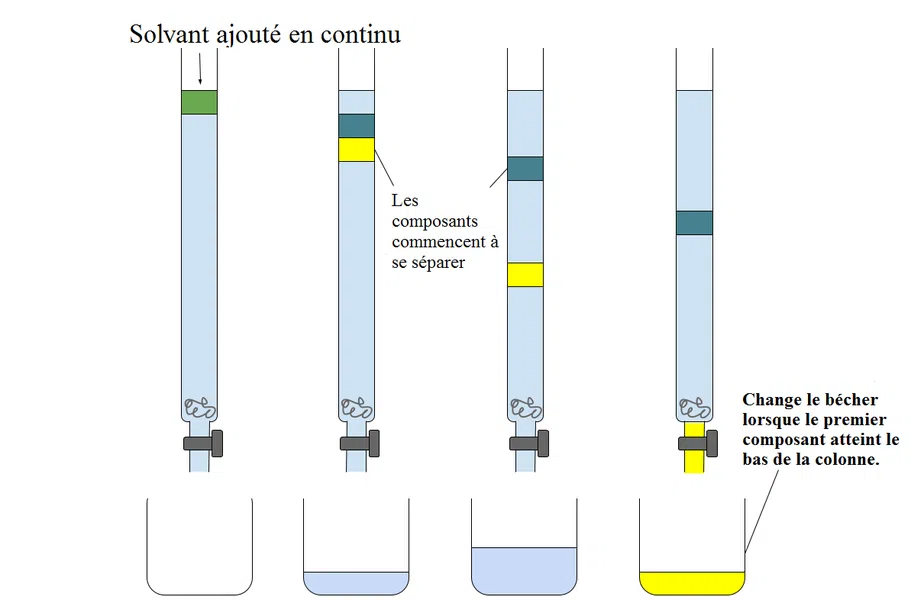
Pourquoi Étudier la Théorie de la Chromatographie ?
Avant d’entrer dans le vif du sujet, posons une question simple : à quoi sert la théorie ?
En pratique, la théorie chromatographique permet de :
- Comprendre pourquoi les composés se séparent dans une colonne.
- Quantifier l’efficacité d’une colonne via des paramètres mesurables.
- Optimiser les conditions opératoires (débit, type de phase, longueur de colonne).
- Prédire la forme des pics et anticiper les problèmes de résolution.
Deux grandes théories coexistent : le modèle statique (théorie des plateaux) et le modèle dynamique (théorie cinétique). Chacun apporte un éclairage complémentaire.
Partie I — La Théorie des Plateaux (Modèle Statique)
1. Origine et Historique
La théorie des plateaux est le modèle le plus ancien de la chromatographie. Elle a été développée par Martin et Synge, qui l’ont adaptée à partir du modèle de Craig (séparation à contre-courant).
L’analogie de départ est celle de la colonne de distillation fractionnée : tout comme une colonne de distillation est divisée en plateaux d’équilibre, la colonne chromatographique est assimilée à une succession de zones fictives appelées plateaux théoriques.
À retenir : Ces plateaux n’ont aucune existence physique réelle. Ce sont des outils mathématiques permettant de modéliser et de mesurer l’efficacité d’une séparation.
2. Principe du Modèle
Dans ce modèle, la colonne est découpée en N plateaux théoriques de hauteur identique H (la HEPT). À chaque plateau :
- Un volume identique de phase stationnaire (dvs) et de phase mobile (dv) sont présents.
- Le soluté échange librement entre les deux phases horizontalement, jusqu’à l’établissement d’un équilibre.
- Il n’y a pas d’échange vertical entre plateaux consécutifs.
La relation d’équilibre entre les concentrations du soluté dans les deux phases est gouvernée par le coefficient de partition K :
K = Cs / Cm
Ce coefficient est une constante thermodynamique qui dépend du soluté, de la nature des phases et de la température.
3. Le Pic Chromatographique et la Courbe de Gauss
À la sortie de la colonne, la variation de concentration du soluté dans la phase mobile est enregistrée sous forme d’un pic chromatographique.
La théorie des plateaux démontre que ce pic suit une distribution de Gauss (courbe en cloche symétrique), caractérisée par :
- Un maximum correspondant au temps de rétention tR.
- Deux points d’inflexion à 60,6 % de la hauteur du pic.
- Une largeur à mi-hauteur δ = 2,354 σ.
- Une largeur à la base ω = 4 σ.
En pratique, les pics réels sont rarement parfaitement gaussiens. Ils peuvent présenter des asymétries (traînée ou front diffus) liées aux isothermes de distribution non linéaires.
4. Les Paramètres Fondamentaux du Chromatogramme
a) Grandeurs de Rétention
Le chromatogramme fournit plusieurs informations clés :
- Temps de rétention (tR) : temps entre l’injection et le sommet du pic. Il sert à l’identification du composé.
- Temps mort (t0 ou tM) : temps de passage d’un composé non retenu. Il reflète le volume mort de la colonne.
- Temps de rétention réduit (t’R = tR – t0) : temps corrigé, plus représentatif de l’interaction réelle soluté-phase stationnaire.
- Volume de rétention (VR = tR × D) : volume de phase mobile nécessaire pour éluer le soluté.
b) Le Facteur de Rétention k (ou k’)
Le facteur de rétention k est l’un des paramètres les plus utiles en chromatographie :
k = QS / QM = (K × VS) / VM
- Si k = 0 : le composé n’est pas retenu par la phase stationnaire.
- Si k augmente : la rétention est plus forte et le temps d’analyse s’allonge.
L’avantage de k sur tR : il est indépendant du débit et de la longueur de la colonne, ce qui facilite la comparaison entre différents systèmes chromatographiques.
5. L’Efficacité de la Colonne : N et HEPT
L’efficacité d’une colonne est mesurée par le nombre de plateaux théoriques N ou sa hauteur équivalente HEPT (H) :
N = tR² / σ² = 16 × (tR / ω)² = 5,54 × (tR / δ)²
H = L / N
- Plus N est grand → plus H est faible → plus la colonne est efficace.
- Un pic fin (σ faible) reflète une haute efficacité et un meilleur pouvoir séparateur.
L’efficacité dépend de plusieurs facteurs :
- La longueur de la colonne (L).
- La qualité du remplissage et la taille des particules.
- Le produit analysé et ses interactions avec les phases.
- La vitesse de la phase mobile (ce point est traité par la théorie cinétique).
6. Sélectivité et Résolution : les Critères de Qualité d’une Séparation
Facteur de Sélectivité α
Le facteur α compare la rétention de deux composés A et B adjacents :
α = kB / kA = KB / KA
- α = 1 → pas de séparation possible.
- Plus α augmente → séparation plus facile.
Facteur de Résolution Rs
La résolution Rs mesure quantitativement la qualité de séparation entre deux pics :
Rs = 2 × (tRB – tRA) / (ωA + ωB)
En pratique :
- Rs ≤ 0,8 → séparation insuffisante.
- Rs = 1 → chevauchement de 2 % des pics.
- Rs ≥ 1,5 → bonne séparation, retour à la ligne de base.
La formule de Purnell relie Rs aux trois paramètres essentiels : N, α et k. C’est un outil précieux pour savoir quel levier actionner pour améliorer une séparation.
7. Limites de la Théorie des Plateaux
Malgré ses apports, ce modèle présente des limites importantes :
- Il suppose que l’équilibre est atteint dans chaque plateau, ce qui n’est jamais le cas en réalité (la phase mobile se déplace en continu).
- Il ne permet pas d’expliquer l’élargissement des bandes en fonction du débit.
- Il ignore la dynamique du système.
C’est pour combler ces lacunes qu’a été développée la théorie cinétique.
Partie II — La Théorie Cinétique (Modèle Dynamique)
1. Introduction : L’Élargissement des Bandes Chromatographiques
La théorie cinétique part d’un constat réel : lors de leur migration dans la colonne, les zones chromatographiques ne restent pas compactes. Elles s’élargissent progressivement.
Cet élargissement est dû à plusieurs phénomènes physiques simultanés, que la théorie cinétique cherche à modéliser et quantifier.
Deux modèles majeurs ont été développés :
- Le modèle de Van Deemter (1956).
- Le modèle de Knox.
2. Le Modèle de Van Deemter
L’Équation de Van Deemter
Proposée en 1956 pour la chromatographie en phase gazeuse (CPG), l’équation de Van Deemter est aujourd’hui l’outil de référence pour optimiser les séparations chromatographiques :
H = A + B/u + C×u
Où :
- H : Hauteur Équivalente à un Plateau Théorique (HEPT).
- u : vitesse linéaire moyenne de la phase mobile (u = L / tM).
- A, B, C : coefficients représentant trois sources distinctes d’élargissement.
Cette équation aide à déterminer le débit optimal de la phase mobile et est fondamentale pour comprendre comment les différents facteurs contribuent à l’élargissement des pics.
Le Terme A — Diffusion Turbulente (Eddy Diffusion)
Le terme A traduit la multiplicité des chemins parcourus par les molécules à travers les espaces inter-particulaires de la colonne.
Certaines molécules empruntent des chemins courts et avancent vite ; d’autres parcourent des trajets plus longs. Cette dispersion est inévitable dans une colonne remplie.
A = 2 λ dp
- λ : qualité du garnissage.
- dp : diamètre des particules.
Le terme A peut être réduit en améliorant le procédé de tassement et en utilisant des particules plus petites et plus uniformes, ce qui minimise la variation des longueurs de trajet.
Exemple concret : Les colonnes HPLC sub-2 µm utilisées en UHPLC (Ultra High Performance Liquid Chromatography) exploitent ce principe : des particules extrêmement petites (1,7 µm au lieu de 5 µm) réduisent considérablement A et améliorent l’efficacité.
Le Terme B — Diffusion Longitudinale
Le terme B traduit la tendance naturelle des molécules à diffuser depuis les zones concentrées vers les zones diluées, dans le sens de la colonne.
B = 2 γ Dm
- Dm : coefficient de diffusion moléculaire dans la phase mobile.
- γ : facteur de tortuosité.
Ce terme est significatif en CPG (les gaz diffusent rapidement) mais quasi négligeable en chromatographie liquide (CPL), où Dm est 10⁴ à 10⁶ fois plus faible.
Le Terme C — Résistance aux Transferts de Masse
Le terme C est souvent le facteur limitant en CPL. Il traduit le délai que mettent les molécules à s’équilibrer entre phase mobile et phase stationnaire.
- Certaines molécules, situées au centre du flux, avancent plus vite.
- Celles en périphérie ou adsorbées sur la phase stationnaire restent en retard.
- Résultat : des molécules identiques se retrouvent à des positions différentes → élargissement du pic.
La Courbe de Van Deemter et la Vitesse Optimale
La courbe H = f(u) est une branche d’hyperbole passant par un minimum. Ce minimum correspond à la vitesse optimale de la phase mobile, celle qui minimise H et maximise l’efficacité.
En pratique :
- À faible vitesse : le terme B (diffusion longitudinale) domine → les pics s’élargissent.
- À grande vitesse : le terme C (transfert de masse) domine → les pics s’élargissent également.
- À la vitesse optimale : l’équilibre entre les trois termes est atteint.
Application industrielle : En HPLC pharmaceutique, les laboratoires ajustent précisément le débit de la phase mobile en s’appuyant sur la courbe de Van Deemter pour maximiser la résolution tout en minimisant le temps d’analyse.
La Loi de Darcy et les Contraintes de Pression
Augmenter la vitesse u a un coût : la pression augmente selon la loi de Darcy. Au-delà de 180 bars (environ), les contraintes mécaniques deviennent problématiques pour les colonnes et les équipements standards.
C’est notamment pour contourner cette limite qu’ont été développées les colonnes UHPLC, capables de travailler jusqu’à 1 000 bars avec des particules sub-2 µm.
3. Le Modèle de Knox
Les Grandeurs Réduites
Le modèle de Knox utilise des grandeurs adimensionnelles (hauteur de plateau réduite h, vitesse réduite ν) normalisées par le diamètre des particules dp. Cela permet de comparer des colonnes de nature et de dimensions très différentes.
L’Équation de Knox
h = B/ν + A ν^(1/3) + C ν
La courbe de Knox passe par un minimum remarquable pour ν = 2,7 et h = 2,4, ce qui constitue un référentiel universel d’efficacité.
Trois régions sont distinguées sur cette courbe :
- Faible ν : la diffusion longitudinale (terme B) domine, efficacité dégradée aux très faibles vitesses.
- Grande ν : le terme C domine, efficacité dégradée aux grandes vitesses.
- Région intermédiaire : le terme A prédomine, passage par le minimum d’efficacité optimale.
Intérêt du Modèle de Knox
Les équations de Van Deemter et de Knox sont les deux outils les plus utilisés pour calculer la hauteur théorique de plateau en fonction de la vitesse de flux. La courbe de Knox offre en plus un cadre universel pour :
- Comparer la chromatographie en phase gazeuse, liquide et supercritique.
- Évaluer des colonnes remplies de particules de différentes tailles et natures.
- Transférer des méthodes chromatographiques entre différents systèmes.
Partie III — Asymétrie des Pics et Isothermes de Distribution
1. Symétrie vs. Asymétrie
Dans des conditions thermodynamiques idéales, les pics sont parfaitement gaussiens. Dans la réalité, des déformations apparaissent, liées à la nature de l’isotherme de distribution (relation Cs = f(Cm)) :
- Isotherme linéaire (Cs = K×Cm) → pic gaussien symétrique. (Cas idéal)
- Isotherme convexe → pic à front diffus (la substance avance trop vite à forte concentration).
- Isotherme concave → pic avec traînée ou “tailing” (la substance est retenue trop longtemps à forte concentration).
L’asymétrie est quantifiée par le facteur de traînée, dont la valeur idéale est 1 (pic parfaitement symétrique). En HPLC pharmaceutique, un facteur de traînée compris entre 0,8 et 1,5 est généralement acceptable.
2. Optimisation de la Résolution : La Formule de Purnell
La formule de Purnell est l’outil ultime de l’optimisation chromatographique. Elle exprime Rs en fonction des trois leviers d’action :
Rs = (√N / 4) × ((α – 1) / α) × (k / (1 + k))
Cela signifie que pour améliorer une séparation, on peut agir sur :
- N (Efficacité) : en augmentant la longueur de colonne, en réduisant la taille des particules, ou en optimisant le débit.
- α (Sélectivité) : en changeant la phase stationnaire, la composition de la phase mobile ou la température.
- k (Rétention) : en modifiant le rapport des solvants dans la phase mobile.
Exemple pratique : En HPLC en phase inverse, passer d’un mélange eau/acétonitrile 50/50 à 65/35 augmente k, améliore α, et peut faire passer Rs de 0,9 (insuffisant) à 1,8 (excellente séparation), sans aucun changement de colonne.
Conclusion : Deux Théories Complémentaires au Service de la Pharmacie
La théorie des plateaux et la théorie cinétique ne s’opposent pas — elles se complètent.
- La théorie des plateaux vous donne les outils pour mesurer et comparer l’efficacité des colonnes, identifier les solutés et évaluer la qualité d’une séparation.
- La théorie cinétique (Van Deemter, Knox) vous explique pourquoi les pics s’élargissent et vous donne les leviers pour y remédier.
Points Clés à Retenir
- HEPT (H) : plus elle est faible, plus la colonne est efficace.
- N : plus il est élevé, meilleur est le pouvoir séparateur.
- α : facteur de sélectivité — indispensable pour que deux composés se séparent.
- Rs ≥ 1,5 : critère de bonne séparation en pratique.
- Vitesse optimale : il existe une vitesse de phase mobile qui minimise H — la courbe de Van Deemter permet de la déterminer.
- Taille des particules : réduire dp améliore simultanément A, la sélectivité et le débit optimal, au prix d’une pression plus élevée.
Articles similaires
Les Comprimés Pharmaceutiques : Tout Comprendre sur la Forme Médicamenteuse la Plus Utilisée au Monde
Les comprimés sont la forme pharmaceutique la plus prescrite et la plus consommée dans le monde entier. Simples d’utilisation, économiques...
Lire l'article
Bioénergétique : Comprendre l’Énergie du Vivant
Introduction : Pourquoi les cellules ont-elles besoin d’énergie ? Chaque cellule vivante — qu’il s’agisse d’un neurone, d’un entérocyte ou...
Lire l'article
Les Champignons Inférieurs : Chytridiomycota et Zygomycota — Classification, Biologie et Importance
Introduction : Qu’est-ce qu’un champignon inférieur ? Les champignons représentent l’un des règnes les plus fascinants du vivant. On en...
Lire l'article