

Chromatographie liquide haute performance
Chromatographie liquide haute performance
Introduction : C’est la première méthode chromatographique décrite (comme méthode non instrumentale). La chromatographie en phase liquide ( CPL) sur colonne est un outil analytique performant utiliséepratiquement tous les composés chimiques .
Ce développement est du aux : -Meilleur compréhension des mécanismes d’interactions : soluté /phase mobile/ phase stationnaire. – grandes efficacités obtenus avec des phases stationnaires de plus en plus fines -progrès importants effectués dans le domaine de l’appareillage, en particulier pour la détection. Historique : 1967 : grâce aux travaux de HUBER et HUZSMAN, le début de la chromatographie liquide à haute vitesse, plus tard appelée à juste titre chromatographie liquide haute performance (CLHP) 1969 : après le 5e Symposium International « Advances in Chromatography » développement rapide de la chromatographie en phase liquide CLHP ou HPLC.
II.
Principe de la chromatographie en phase liquide :
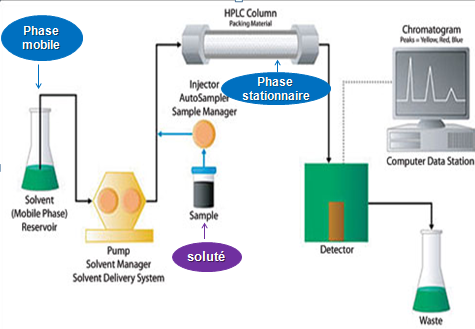
III.
Principaux modes en chromatographie liquide : Il existe plusieurs modes possibles en chromatographie liquide à haute performance.ils correspondent chacun à un type précis d’interaction : -Adsorption – Partage – échange d’ion -exclusion par taille
1.
Chromatographie d’adsorption: liquide-solide : -Adsorption est la fixation d’un liquide ou d’un gaz sur un support solide – Elle doit être réversible par désorption par l’éluant qui remplace le soluté-Interaction L’éluant – soluté : polarités, de structure chimique voisines
La phase stationnaire est un solide adsorbant, la silice et l’alumine sont les phases stationnaires les plus utilisées. Le soluté et le solvant peuvent être attirés par les sites polaires situés à la surface de la phase stationnaire adsorbante. Les solutés présentent des interactions différentes vis-à-vis de la phase stationnaire leur séparation est possible
Groupement silanols Si-OH à la surface de la silice
2.
Chromatographie de partage : la technique de chromatographie liquide la plus utilisée. -Le partage de solutés entre deux phases non miscibles (rétention en fonction de la solubilité relative) -les espèces les plus retenues sont celles ayant une plus grande affinité (solubilité) pour la phase stationnaire, relativement à la phase mobile(éluant). – Éluants immiscibles à la phase stationnaire et compatible avec les détecteurs. -La phase stationnaire est un liquide déposé ou greffé sur un support solide.
Il y a 2 types de chromatographie de partage: – Chromatographie de partage sur phase normale (NLC). -Chromatographie de partage sur phase inversée(RPLC).
| Phase inverse | Phase normale (classique) | |
| Phase stationnaire | Non-polaire(Hydrophobe)e.g.-silice greffée par une chaîne alkyle (C8, C18)-phényle (RP-2, RP-8, RP-18)-Si-(CH2)nCH3 | Polaire (Hydrophile)– silice-C2H4CN–amine polaire; nitriles-diol |
| Phase mobile | Polaire–eau-méthanol-tétrahydrofuranne | Non polairee.g.-n-hexane-chloroforme |
Rappel sur la polarité des solvants: hydrocarbures ˂éthers ˂esters˂ cétones˂ aldéhydes˂ amides ˂amines˂ alcools˂ H2O˂phénol˂acides.
A/Mécanisme de séparation NLC : a- Une solvatation des greffons (soluté) polaires par le solvant le plus polaire de la phase mobile. b- Les molécules de solutés vont : -Soit interagir avec la phase liquide polaire stationnaire. -Soit déplacer les molécules de phase liquide stationnaire et de façon d’autant plus intense que leur polarité est plus élevée. Pour une phase mobile donnée, plus le soluté est polaire plus il est retenu. Phase mobile en NLC : Le pouvoir élutif augmente avec la polarité du solvant : Solvant polaire (très élutif) → solvant fort. Solvant apolaire (peu élutif) → solvant faible.
| Exemples | Solvant |
| Ether isopropyliqueChloroformeDichloroéthaneChlorure de méthylène | FortSélectif |
| Alcanes (hexane)(pour ajuster K’) | FaibleNon sélectif |
Pour améliorer la sélectivité, il faut, tout en gardant le même pouvoir élutif de la phase mobile : Pour les solutés non ionisables : changement du solvant fort (modification des interactions). Pour les solutés ionisables : la phase aqueuse doit être tamponnée à un pH adéquat. B/Chromatographie de partage sur phase inversée(RPLC) : 80% des séparations en CL sont effectuées en phase inversée: Deux mécanismes de rétention ont été proposés : -Soluté moyennement apolaire: 1.Une fixation du mélange (eau-solvant organique) à la surface des greffons apolaires. 2. un partage des solutés entre la phase mobile et la phase liquide adsorbée. –Soluté très apolaire La rétention une conséquence de l’effet hydrophobe : il y’aura Une compétition entre le soluté et les molécules du solvant organique de la phase mobile. L’importance relative de ces deux mécanismes dépend de la nature du soluté, en particulier de leur caractère hydrophobe. Pour une phase mobile donnée, plus le soluté est polaire moins il est retenu.
Phase mobile en RPLC : Le pouvoir élutif diminue avec la polarité du solvant.
Plus le solvant est polaire, moins il est élutif : Solvant polaire (peu élutif) → solvant faible. Solvant apolaire (très élutif) → solvant fort. Le pouvoir élutif souhaité est obtenu pour des valeurs de K’ comprises entre 1 et 10. Pour améliorer la sélectivité, il faut, tout en gardant le même pouvoir élutif de la phase mobile : Pour les solutés non ionisables : changement du solvant fort (modification des interactions). Pour les solutés ionisables : la phase aqueuse doit être tamponnée à un pH adéquat.
| Pouvoir éluant | solvant |
| 4,4 | THF |
| 4,2 | Isopropanol |
| 3,6 | Ethanol |
| 3,1 | Acétonitrile |
| 3,0 | Méthanol |
| 0.0 | Eau |
Solvants utilisés en phase inversée:
MeOH :caractère acide
Éthanol:caractère acide
CH3CN:caractère basique
THF: caractère polaire
H20(pour ajuster K’)
Exemple d’optimisation d’une séparation de chromatographie en phase inversée: Phase mobile :4 solvants utilisés pour optimiser α et garder K’ fixe : (Méthanol ,Acétonitrile ,Tétrahydrofurane ,H2O) On évaluer chaque solvant seul avec H2O pour optimiser K’ (Selon les tR) selon l’équation suivante:P’AB = ΦAP’A + ΦBP’B et
Φ : proportion du volume, P’ : indice de polarité, k’ : facteur de rétention du soluté
En phase normale :
PM : MeOH 71% , H2O 29%
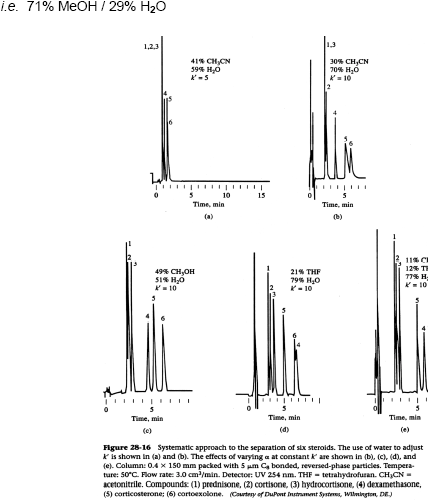
Figure 1: optimisation de séparation de six stéroïdes par CL en phase inversée
Comparaison entre les techniques de la chromatographie de partage
| Technique | Avantage |
| RPLCChromatographie de partage en phase inversée | -facile -Séparation des analytes polaires (solubles ou non solubles dans l’eau) – permet de contrôler : pH,mélange des solvants organiques , la concentration. |
| NLCChromatographie de partage en phase normale | – Séparation des substancesinsolubles ou dégradables dans l’eau -Séparation des solutés très hydrophobes (apolaires) –Séparation des isomères |
3.
Chromatographie par exclusion (SEC) : Principe: Molécules séparées par différence de taille et non par phénomène d’affinité car séparation indépendante de la nature du solvants (seulement véhicule ) Plusieurs appellations selon la nature du support:–Chromatographie par filtration sur gel( support hydrophile) : séparation des protéines, peptides. -Chromatographie par perméation de gel (support hydrophobe: organique) Phase stationnaire (Phase dispersée) Un gel ,structure en tamis permet le criblage des molécules selon la taillePhase mobile(phase dispersante) induit le gonflement de la PS et dissout l’échantillon: Eau pour les gels hydrophiles. Solvant organiques (chloroforme, DMF…) si le gel est hydrophobe.
Caractéristiques de la chromatographie sur gel: -toutes les molécules sont éluées. – temps de séparation prévisible car correspond au volume d’élution total. -volume d’élution est en fonction de la taille des molécules, d’ou détermination Mr (masse relative) deux molécules peuvent être séparé s’il existe au moins 10% de différence de Mr. -Les espèces ayant une dimension importante auront un trajet plus court dans la colonne et seront éluées en premier. -l’efficacité, est en général inferieure à la chromatographie liquide habituelle.
4.
Chromatographie d’échange d’ions : Principe: -Séparation des espèces chargées, des mélanges d’anions ou des cations par résines échangeuses d’anions ou des cations. Phase stationnaire: Insoluble dans l’eau, stable chimiquement, ses particules sphériques et uniformes. Le support échangeur le plus utilisé la résine synthétique comportant un contre ion « ion actif » peut être échangé réversiblement avec des « ions mobiles » de même charge. Phase mobile: solution aqueuse ou un mélange (eau –méthanol)
| Échangeurs cationiques | Type | Nature de résine |
| polymère anionique(Groupement fonctionnel) | -carboxylique-sulfonates-phosphorique-hydroxylique-aminodiacétique | |
| cation actif | -Proton-ion métallique | -Résine acide-Résine chélatante |
| Échangeurs anioniques | type | Nature de résine |
| Polymère cationique(Groupement fonctionnel) | Bases libres ,oxydes de NH4+Sels deNH4+ | |
| Anion actif | -OH–-anions des sels | – Résine basique |
figure 2 : Classification des groupements fonctionnels des échangeurs d’ions
Loi de DARCY Exprime le débit d’un fluide incompressible filtrant au travers d’un milieu poreux entre deux points.
Δp = Pe – Ps : perte de charge ( pa) / Pe pression d’entrée, Ps pression de sortie L: longueur de colonne (m) ɳ: viscosité de la PM (pa.s) Φ: résistance à l’écoulement Dp2 : diamètre des particules de la Ps IV/Appareillage :1.Réservoirs de phase mobile : Ces réservoirs, généralement en verre, en nombre de quatre, doivent être :
Etanches pour éviter l’évaporation des solvants, la contamination par l’eau atmosphérique et dissolution de l’oxygène de l’air.
Equipés d’un dispositif qui chasse les gaz dissous : barbotage de l’azote, agitation mécanique, etc.
D’une contenance : – 1à 2 litres pour la chromatographie analytique. – Jusqu’à 10 litres pour la chromatographie préparative.
2.
Système de pompage : Caractéristiques : Une pompe idéale doit réunir les qualités suivantes : –Obtention de grandes pressions (jusqu’à 600 bars). –Débit constant, reproductible et sans pulsation. –Grandes gammes de débit ( de 0.1 à 10 ml/mn) . –Résistance à la corrosion par les solvants. -Faible volume de la chambre de la pompe. –Délivrance non limitée du solvant.
Différents types de pompes : a)Pompe à pression constante (pompe pneumatique):Principe : ce sont des pompes qui, par l’intermédiaire d’un gaz inerte, exerce une pression constante à la phase mobile. Avantages : -Obtention de débit non pulsé. -Moins coûteuses.Inconvénients : –Impossibilité d’obtenir des débits élevés. -Le volume de solvant délivré est limité. -Ne conviennent pas au système de gradients d’élution. Remarque : ces pompes sont réservées actuellement pour le remplissage des colonnes. b) Pompes à débit constant : b1) Seringue commandée par un vis: Principe : un piston se déplace à l’aide d’une vis sans fin actionné par un moteur pas à pas, refoulant ainsi la phase mobile. Avantages : –Obtention de débits élevés. -Écoulement sans pulsation.
Inconvénients : –Délivrance de solvant limité (environ 250 ml). -Mal adapté aux changements des solvants.
b2)Pompe alternative: Principe : elle est constituée d’une petite chambre qui se remplit et se vide en fonction d’un mouvement va-et-vient d’un piston. Avantages : -Obtention de débits élevés. -Délivrance de solvant non limité. -Faible volume de la chambre de piston. -Adaptable au régime de gradient d’élution. Inconvénients : -Écoulement pulsé. -Pour remédier ce problème, on utilise actuellement des pompes à deux tètes montées en opposition et à came en forme cardioïde permettant d’obtenir un débit totale de la phase mobile sans pulsation.
Élution :
A/Mode isocratique: –La composition de l’éluant reste constante durant le temps de l’analyse – Utilisée pour les séparations simples
B/Mode gradient: -La composition de l’éluant augmente avec le temps de l’analyse -utilisée pour la séparation de mélanges complexes. -Utilisée pour les méthodes de développement cas des mélanges de nature inconnue.
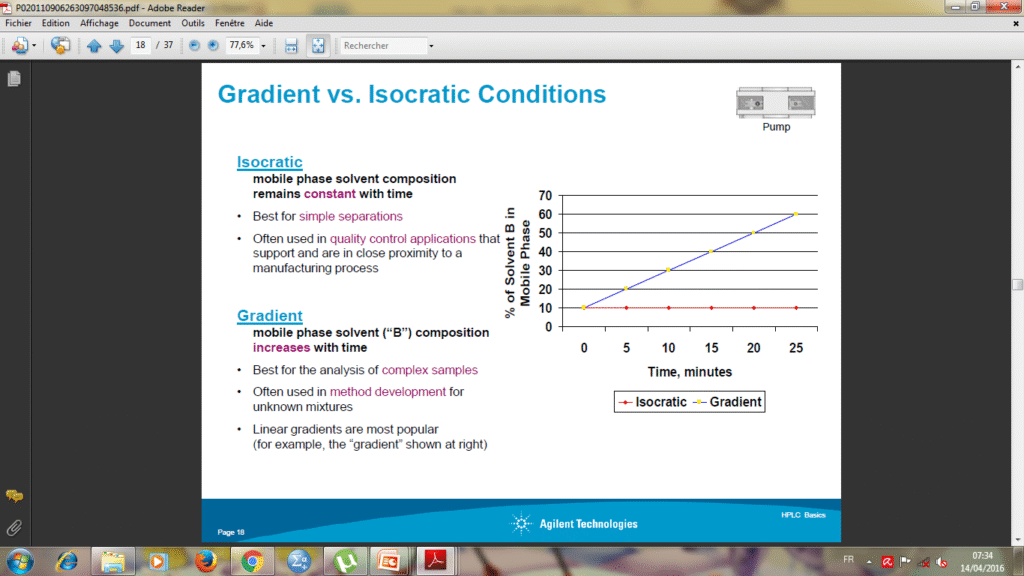
Les gradients peuvent être réalisés de deux manières : En amant de la pompe : dans les chromatographes à une seule pompe, le mélange des solvants est obtenu en programmant le temps d’ouverture des vannes proportionnelles. En aval des pompes : dans les chromatographes à deux ou trois pompes, le gradient est obtenu ; en modifiant à débit total constant, le rapport des débits des pompe
3.
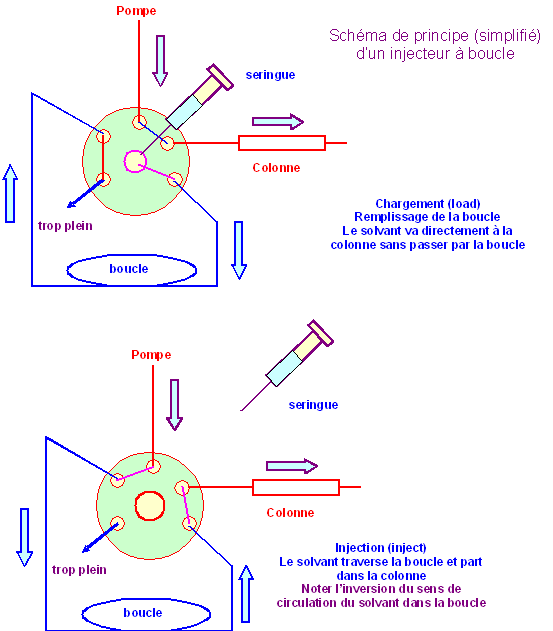
Les injecteurs : Les procédés d’injection peuvent être regroupés en deux catégories : 3.a) Procédés par injection directe : Ces procédés sont applicables lorsque : – Les quantités injectés sont peu importantes. – Les pressions inférieures à 150 bars. 3.b) Procédés par boucle d’échantillonnage : Ils peuvent fonctionnés sous fortes pressions (les plus utilisées).
4.
Colonnes : En acier inoxydable, en titane peut être recouverte de verre. Les colonnes sont généralement : -De 5 à 25 cm de longueur. -De 4 à 5 mm de diamètre intérieur. -Il existe des microcolones de 3cm à 7,5 cm de longueur
Les précolonnes :
Colonne complexante : -Placé entre le réservoir de la phase mobile et l’injecteur. – Le solvant est dissout partiellement les particules de silice avant d’entrer dans la colonne analytique. -la saturation du solvant minimise les pertes de la colonne analytique
Colonne de garde : –Placé entre l’injecteur et la colonne analytique-Elle est remplie avec la même phase stationnaire que la colonne analytique. -Cette colonne empêche les impuretés d’atteindre la colonne. 5.
Détecteurs : Un instrument placé immédiatement à la sortie de la colonne, et qui permet de suivre en continu la séparation et de mesurer la concentration des solutés. A)Caractéristiques : -donner une réponse stable, rapide et reproductible. -garantir une bonne sensibilité de détection. -ne pas altérer la qualité de la séparation. B) classification des détecteurs : On peut classer les détecteurs soit selon leur principe de mesure soit selon le degré d’information qu’ils peuvent apporter. Dans le premier classement, on distingue : Détection directe : mis en œuvre d’une propriété spécifique du soluté. Détection indirecte : mis en œuvre d’une propriété spécifique d’un des constituants de l’éluant. Détection différentielle : mesure une différence de propriété entre le soluté et l’éluant.
| Classement des Détecteurs selon leur principe de mesure | ||
| Détection directe | Détection indirecte | Détection différentielle |
| Spectrophotomètre UV et IR Fluorimètre Détecteur éléctrochimique Diffusion de la lumière Polarimètre Radioactivité Spectromètre de masse Spectromètre de résonance magnétique nucléaire Ionisation et émission de flamme | Spectrophotomètre UV (mesure de la diminution de l’absorbance)Fluorimètre (mesure de la diminution de l’absorbance) | Réfractomètre ConductImètre Permitivité |
Dans le deuxième classement, on regroupe : -Détecteur simple : qui assure la visualisation du chromatogramme. -Détecteur semi-informatif : fournit des critères de pureté et permet d’optimiser la sélectivité. -Détecteur intelligent : fournit des éléments d’identification.
| Classement des Détecteurs selon le degré d’information fournie | ||
| Détecteurs simples | Détecteurs semi-informatifs | Détecteurs intelligent |
| Spectrophotomètre UV-Visible Fluorimètre Réfractomètre Détecteur éléctrochimique Conductomètre Radioactivité Diffusion de la lumière Ionisation et émission de flamme Absorption atomique | Spectrophotomètre UV-Visible programmable ( temps et longueur d’onde) Spectrophotomètre à deux longueurs d’onde Fluorimètre programmable ( temps et longueur d’onde) Détecteur éléctrochimique programmable et à plusieurs électrodes indicatrices. Pomarimètre | Spectromètre de masse.Spectrophotomètre à barrette de diodes.Spectrophotomètre infrarouge à transformée de Fourier.Spectromètre de résonance magnétique nucléaire. |
5.1) Réfractomètre différentiel : Principe : le réfractomètre différentiel mesure en continu la différence d’indice de réfraction entre la phase mobile et l’effluent de la colonne chromatographique.
En effet, la réponse R1 du réfractomètre est donnée par la relation :
R1 = Z (n – n0)
Avec Z : constante caractéristique de l’appareil. n : indice de réfraction de l’effluent. n0 : indice de réfraction de la phase mobile
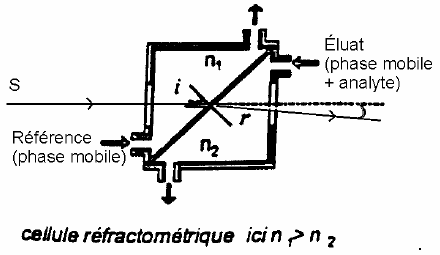
Caractéristiques : -Non destructif. -Universel.La réponse dépend à la fois de la nature du soluté et de la phase mobile. -Quantité minimale détectable est de l’ordre : 100 à 200 ng. -Grande sensibilité aux variations de la température et de la pression (réfractomètre ultra-thermostaté). -Ne peut pas être utilisé en mode gradient. 5.2) Détecteur à diffusion de la lumière (DDL) : Principe : le principe est fondé sur l’évaporation partielle de l’effluent de la colonne de façon à obtenir un brouillard de particules solides ou liquides du soluté qui traverse un faisceau lumineux. La lumière diffusée sous un ongle déterminé est détecté par un photomultiplicateur.Caractéristiques : -Non destructif. -Universel. -Sa sensibilité varie peu en fonction de la nature du soluté. -Compatible avec le gradient d’élution. -La mise en œuvre de ce détecteur est limité à l’emploie de phases mobiles volatiles et de solutés non volatils. 5.3) Spectrophotomètre UV-Visible: Principe: la réponse de ce type de détecteur est fondée sur la loi de Beer-Lambert : A = log (I0 / I) =ξ LC Avec ξ : absorptivité molaire du soluté à la longueur d’onde λ . I0 : intensité du rayonnement incident. I : intensité du rayonnement transmis. L : longueur du chemin optique. C : concentration du soluté dans l’effluent. Caractéristiques : -Sélectif. -Non destructif. -La détectabilité de ce détecteur dépend essentiellement de l’absorptivité ξ du soluté et, par conséquent, de sa nature. -Pour une valeur moyenne de ξ, les quantités minimales détectables sont de l’ordre de ng. -Quantification se fait après étalonnage préalable. -Peu sensible à la température et au débit. -Peut être utilisé en mode gradient. -Limité par le “cut off” de la phase mobile. Variantes : -Spectrophotomètre dont la longueur d’onde est réglée par l’opérateur. -Spectrophotomètre programmable en longueur d’onde dans le temps -Spectrophotomètre à barrette de diodes : permet une saisie en trois dimensionsabsorbance-longueur d’onde-temps.
L’identification ou la mise en œuvre des critères de pureté des solutés se font par comparaison de leurs spectres UV avec ceux stockés dans une bibliothèque.
5.4) Spectrofluorimètre : Principe : certains composés organiques absorbent des radiations dans l’UV et réémettent une fraction de la lumière absorbée à une longueur d’onde supérieure. La loi fondamentale de la fluorimétrie est :
If = K Iabs = K (I0 – I)
Avec If : intensité de la lumière émise par fluorescence. K : facteur de proportionnalité.
Caractéristiques : -Non destructif. -Très sélectif (Des techniques de dérivation permettent d’élargir le champ d’application). -La réponse n’est linéaire qu’aux faibles concentrations. -La sensibilité est supérieure à celle de l’absorptiomètre. -Quantité minimale détectable est de l’ordre de fentomoles(analyse des traces). Variantes : -Les spectrofluorimètres munis de monochromateur à excitation et à émission. -Les spectrofluorimètres munis de monochromateur à excitation et un filtre à l’émission. -Les spectrofluorimètres à commutation de gammes automatiques programmables en longueurs d’onde dans le temps. 5.5) Détecteur électrochimique : Principe : considérons une substance élecroactive contenue dans un liquide en mouvement au contact d’une électrode, la charge Q échangée avec l’électrode est donnée par la loi de Faraday : Q = ne F N Avec ne : nombre d’électron mis en jeu. F : constante de Faraday. N : nombre de moles du soluté. Le courant « it » qui circule dans la cellule d’électrolyse à l’instant t est :
it = ne F (Ci – Cf) D Avec Ci : concentration du soluté à l’entré de la cellule. Cf : concentration du soluté à la sortie de la cellule. D : débit de l’effluent à travers la cellule. Caractéristiques : –Destructif. -Sélectif (substances électroactives). -La sensibilité dépend essentiellement du choix du potentiel d’électrode. -Les phases mobiles utilisées doivent être suffisamment conductrices pour permettre le passage du courant. -Interférence de l’oxygène dissout dont l’élimination est très difficile.