

LES GENERALITES SUR LES METHODES CHROMATOGRAPHIQUES
INTRODUCTION
Chromatographie est un terme général, utilisé pour définir des méthodes de séparation basées sur la distribution d’un soluté entre deux phases, l’une étant mobile (un gaz ou un liquide), l’autre stationnaire (un solide ou un liquide).
C’est en 1906 à la suite des travaux de Tswett, que naquit la méthode appelée chromatographie (du grec chroma= couleur).
Il déposa dans une colonne remplie de carbonate de calcium finement pulvérisé un mélange de pigments végétaux (chlorophylles et xanthophylles) dissous dans de l’éther de pétrole.
Ceux-ci s’adsorbaient au sommet de la colonne. L’ajout continu de ce solvant pur au sommet de la colonne provoqua la migration, du haut vers le bas, de chaque pigment à une vitesse qui leur était propre.
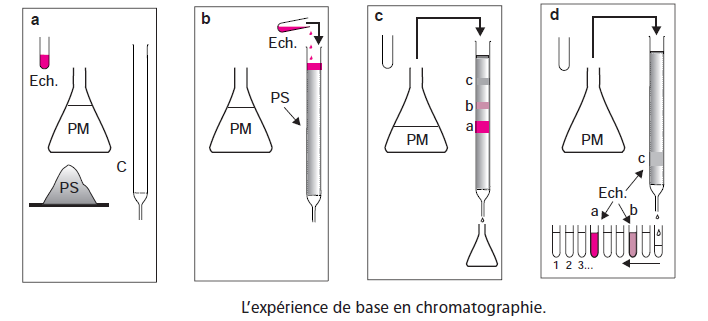
II. DÉFINITIONS
La chromatographie est un procédé physico-chimique de séparation des constituants d’un mélange homogène ou hétérogène liquide ou gazeux.
Les mélanges hétérogènes ou sous forme solide peuvent être mis en solution par emploi d’un solvant.Elle peut être analytique ou préparative.
Phase stationnaire: matériau emprisonné dans une colonne ou fixée sur un support (solide ou liquide)
Phase mobile (éluant): fluide qui se déplace au contact de la première avec une vitesse déterminée
( gaz ou liquide)
Élution : passage d’un composé sur une colonne
Éluat : solution recueillie à la base de la colonne
Chromatogramme : enregistrement des pics perçus par le détecteur
Chromatographe : appareil qui rassemble autour d’une colonne tout un ensemble d’accessoires destinés à assurer la répétabilité des expériences
III.PRINCIPE
Séparations sont basées sur les différences de distribution de solutés entre deux phases non miscibles ( suite continue d’un nombre considérable d’équilibres) Déplacement des composés dans la colonne selon une vitesse différente en fonction de leur affinité pour les deux phases.
Coefficient de distribution (ou de partage) du soluté entre la phase stationnaire et la phase mobile
K = CS/CM (caractéristique du soluté)
CS = concentration dans la phase stationnaire
CM = concentration du soluté dans la phase mobile
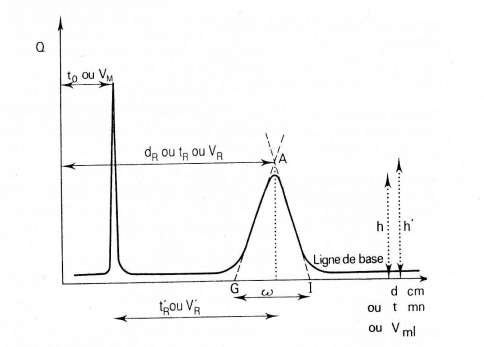
Dans le cas de la chromatographie sur colonne on obtient, à la sortie du détecteur, un « signal » : le chromatogramme
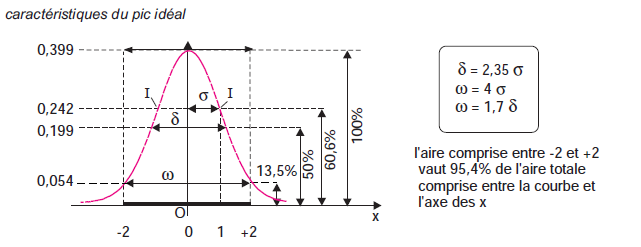
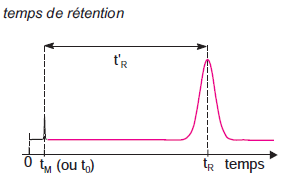
Un constituant est caractérisé par son temps de rétention tR, qui représente le temps écoulé entre l’instant de l’injection et celui qui correspond sur le chromatogramme au maximum du pic qui lui est lié.
Dans le cas idéal tR est indépendant de la quantité injectée.Un constituant non retenu sort de la colonne au temps tM, appelé temps mort (désigné également par t0).
IV.CLASSIFICATION DES MÉTHODES CHROMATOGRAPHIQUES
Selon le phénomène physico-chimique
Chromatographie liquide/solide (ou d’adsorption):
– La phase stationnaire est un milieu solide
– Due à des forces de cohésion (forces de Van der Waals, interactions polaires)
– Carbonates de calcium ou l’inuline (un polymère en poudre très fine du sucre ordinaire).
Chromatographie liquide/liquide ou de partage (CLL):
La phase stationnaire est un liquide
Immobilisé sur un matériau inerte et poreux qui n’a qu’un rôle de support.
Due à la différence de solubilité
Chromatographie ionique:
– La phase stationnaire solide comporte en surface des sites ioniques
La phase mobile est une solution-tampon aqueuse de sels d’acides ou de bases.
Séparation due à des échanges entre les ions de l’échantillon avec ceux de la phase stationnaire.
Coefficients de distribution ionique
Chromatographie d’exclusion-diffusion:
La phase stationnaire est un matériau comportant des pores dont les dimensions sont choisies en rapport avec la taille des espèces à séparer.
Selon la nature, aqueuse ou organique de la phase mobile, cette technique est désignée par filtration sur gel ou perméation de gel.
Le coefficient de distribution prend le nom de coefficient de diffusion
Chromatographie de paires d’ions :
Formation de paires d’ions entre les constituants (ionisés) du mélange à séparer et un contre-ion convenable.
Distribution entre la phase mobile et la phase stationnaire (le plus souvent celle-ci est une phase greffée).
Chromatographie d’échange de ligands :
– La phase stationnaire contient une espèce fixée irréversiblement capable de former des complexes avec les solutés à séparer.
Chromatographie d’affinité :
S’apparente à celle de l’échange de ligand.
Interactions sont connues et spécifiques
Dite bio-spécifique lorsque la phase stationnaire est constituée d’un ligand présentant une grande affinité pour certaines molécules bioactives.
Selon le procédé utilisé
Selon les modalités adoptées pour immobiliser la phase stationnaire on a:
Chromatographie sur colonne
Chromatographie sur couche mince
Chromatographie sur papier
Selon les modalités de migration on a:
Chromatographie d’élution
Chromatographie de développement
Selon La nature des deux phases
Chromatographie en phase liquide (CL)
Chromatographie liquide- liquide
Chromatographie liquide-solide
Chromatographie en phase gazeuse (CG)
Chromatographie gaz-liquide
Chromatographie gaz-solide
Chromatographie en phase supercritique (CS)
V.
ETUDE THÉORIQUE DU PHÉNOMÈNE CHROMATOGRAPHIQUE
V.1.LA THÉORIE DES PLATEAUX
On assimile la colonne chromatographique de longueur L à une succession de plateaux fictifs appelés plateaux théoriques.
Une colonne est constituée de N plateaux théoriques de même hauteur H appelée Hauteur équivalente à un plateau théorique(HEPT) contenant tous les mêmes proportions de PM et PS.
Les équilibres successifs sont à la base de la notion de plateau théorique selon lequel La concentration du soluté dans la phase mobile est en équilibre avec la concentration dans la phase stationnaire de ce soluté.
À chaque nouvel équilibre le soluté a progressé d’un petit disque supplémentaire dans la colonne
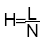
Grandeurs de retention:
Vitesses linéaires moyennes:
Vitesse moyenne linéaire de la phase mobile:
Vitesse moyenne linéaire du soluté:
Fraction de temps dans la phase mobile:
Facteur de rétention (capacité):
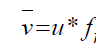
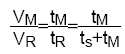
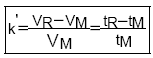
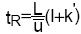
k’ = 0 = pas retenu
k’ entre 1 et 10 mais usuellement entre 2 et 6 k’=2 recommandé par ICH pour éviter l’élargissement des pics
Facteur de séparation (ou sélectivité) entre deux solutés:
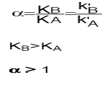
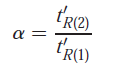
Efficacité d’une colonne
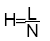
Facteur de résolution entre deux pics :
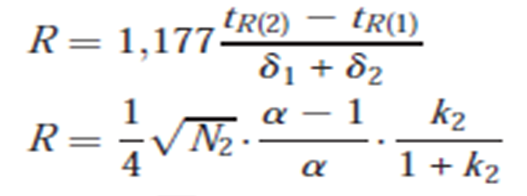
La résolution est une mesure de la qualité d’une séparation chromatographique.
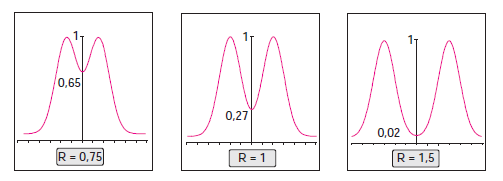
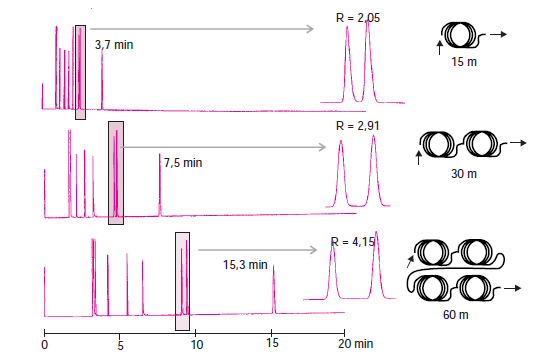
R=1,5 séparation complète
VI.
OPTIMISATION DE LA SÉPARATION:
1.
Rôle du facteur de capacité :
Le facteur de capacité doit être compris entre 1 et 10 pour que son influence soit importante sur la résolution.
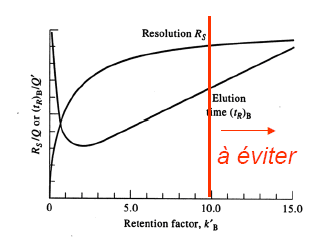
-k’ faible: composés peu retenu.
-k’ fort: composés très retenu.
2.
Rôle de la sélectivité :
3.
Rôle de l’efficacité de la colonne N (ou H) :
N W Rétrécir les pics et éviter le chevauchement
Allonger la colonne Diminuer H
Théorie cinétique
THÉORIE CINÉTIQUE
La théorie des plateaux néglige donc le fait que la chromatographie est un phénomène dynamique résultant du passage continu de la phase mobile sur la phase stationnaire.
En effet, elle ne tient pas compte d’un élargissement ou au contraire d’un rétrécissement des pics que l’on peut pourtant observer pratiquement selon le débit de la phase mobile.
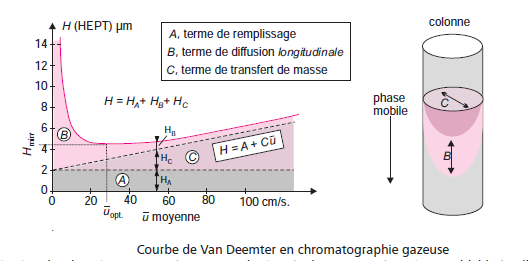
L’influence de la vitesse de la phase mobile a été mise en évidence par Van Deemter qui a proposé la première équation cinétique, dans le cas des colonnes remplies en chromatographie en phase gazeuse :
(A) terme de remplissage:
L’anisotropie d’écoulement du fluide est souvent qualifié de « diffusion turbulente » « facteur de diffusion d’Eddy ». Ce phénomène tire son origine du contournement irrégulier des particules du support solide par le fluide.
Il entraîne une inégalité dans la vitesse de déplacement axial des différents « filets du fluide » qui contourne les grains.
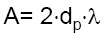
(B)Diffusion moléculaire longitudinale :
La diffusion moléculaire longitudinale est bien connu pour toute particule au sein d’un fluide en mouvement ou non (dispersion par diffusion due à l’entropie du soluté des zones de grande concentration dans des régions de faible concentration, selon les lois de Fick).
Cette diffusion est d’autant plus importante que la vitesse de passage du fluide est plus faible.
L’influence de ce phénomène sur la hauteur d’un plateau est représentée par la relation :
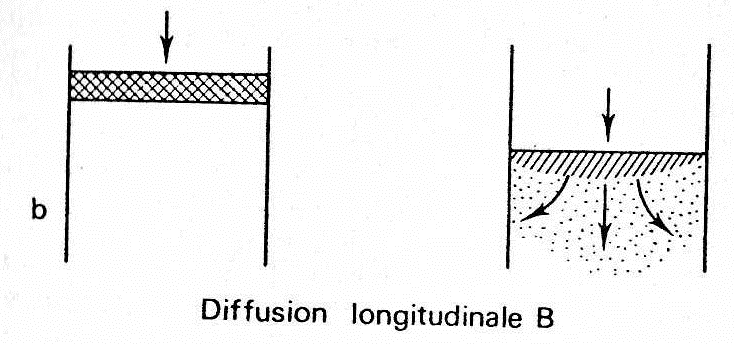
(C) Résistances au transfert de masse
Ce terme C, dû à la résistance au transfert de masse du soluté entre les deux phases, devient prépondérant lorsque le passage est trop rapide pour que l’équilibre soit atteint.
Des turbulences locales au sein de la phase mobile et des gradients de concentration ont pour conséquence de retarder la mise en équilibre (CS ⇔ CM).
La diffusion entre les deux phases n’est pas instantanée, si bien que le soluté sera entraîné hors équilibre.
Il n’existe pas de formule simple rendant compte des différents facteurs intégrés dans le paramètre C.
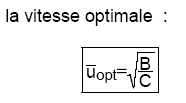
pour réduire w (H):
– Diminuer lesdistances à parcourir dans chaque phase en réduisant le diamètre des particules.
– Diminuer l’épaisseurde PS. – Utiliser une colonne régulièrement remplie et bien tassée.
– Diminuer la température en CPG afin de réduire le coefficient de diffusion (pastrès utile en CL puisque DM est déjà trop petit).
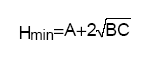
Remarque 1:
L’équation de Van Deemter peut être également, par extension, appliquée à la chromatographie liquide (CL).
Si la formule générale reste la même, la courbe représentative est cependant différente car on n’observe apparemment pas de valeur minimale pour H.Celle-ci croît en effet régulièrement avec le débit ce qui est dû au fait que dans les liquides le terme B est très petit et qu’il n’intervient que pour des valeurs de u très faibles, pratiquement jamais réalisées.
Remarque 2 :
Les chromatogrammes réels sont quelquefois loin de présenter des pics d’aspect gaussien Il y a plusieurs raisons à cela.
En particulier il se produit une irrégularité de concentration dans la zone de dépôt de la substance en tête de colonne.
De plus, la vitesse de la phase mobile est nulle au niveau de la paroi et maximum au centre de la colonne.
L’asymétrie observée d’un pic est traduite par deux paramètres appelés facteur d’asymétrie (Fa) et facteur de traînée (Ft), mesurés à 10 % de sa hauteur.
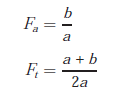
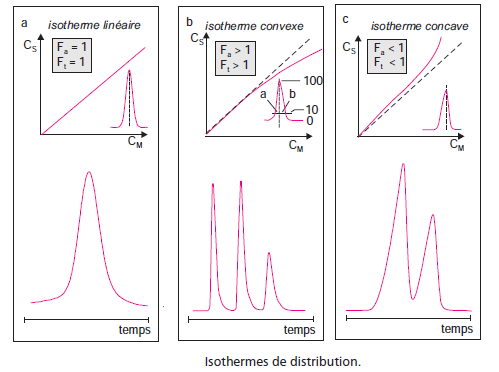
a) Situation idéale correspondant à l’invariance de l’isotherme de concentration
b) Situation dans laquelle la phase stationnaire est saturée – de ce fait la montée du pic est plus rapide que la descente (facteur de traînée plus grand que 1)
c) situation inversée : le constituant est trop retenu dans la phase stationnaire, le temps de rétention est allongé et la montée du pic est plus lente que la descente, qui apparaît normale
VII.
APPLICATIONS
ANALYSE QUALITATIVE:
L’identification d’un composé par chromatographie correspond à une méthode comparative.
Comparaison du temps de rétention à un composé de référence dans les mêmes conditions expérimentales.
Utilise K ‘ou α par l’ajout d’un autre composé dont on connait la sélectivité p/r au premier
associer deux techniques complémentaires par exemple, un chromatographe et un second appareil « en ligne », tel un spectromètre de masse ou un spectromètre infrarouge
Usage des indices de rétention exp: Kovats
ANALYSE QUANTITATIVE
Pour un réglage donné de l’appareil, on admet qu’il existe pour chaque pic du chromatogramme une relation linéaire entre son aire (ou sa hauteur) et la quantité du composé responsable de ce pic dans l’échantillon injecté.
Cette relation est valable pour une plage de concentrations qui dépend du détecteur employé.
On traduit cette hypothèse par :
mi = Ki Ai
1.
MÉTHODE DE L’ÉTALONNAGE EXTERNE
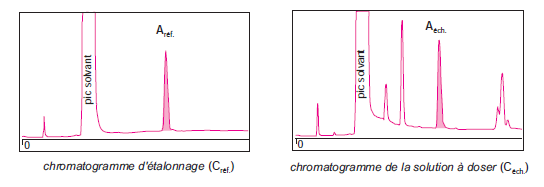
Cette méthode permet de calculer la teneur d’un ou plusieurs constituants apparaissant séparés sur le chromatogramme. Le procédé repose sur la comparaison de deux chromatogrammes obtenus successivement sans changer les conditions de réglage de l’appareil .
Le premier est un chromatogramme de référence acquis à partir d’une solution de référence dans un solvant, du composé qui fait l’objet du dosage.
On injecte un volume V de cette solution et on repère sur le chromatogramme l’aire du pic correspondant.
Le second résulte de l’injection d’un volume identique V de l’échantillon en solution, contenant le composé à doser .
Puisque les volumes injectés sont égaux, il y a proportionnalité entre les aires, qui dépendent des masses injectées, et les concentrations correspondantes :
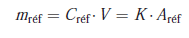
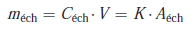
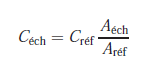
2.
MÉTHODE DE L’ÉTALONNAGE INTERNE (ÉTALON INTERNE)
Cette deuxième méthode repose sur l’utilisation du coefficient de réponse relatif de chaque composé à doser vis-à-vis d’un marqueur introduit comme référence.
Cela permet de s’affranchir de l’imprécision concernant les volumes injectés, un handicap de la précédente méthode.
Les aires des pics des produits à quantifier sont comparées avec celle d’un composé de référence, appelé étalon interne, introduit à une concentration connue dans l’échantillon.
Il doit être pur et ne pas se trouver initialement dans l’échantillon
Son pic d’élution doit être bien résolu par rapport à tous ceux qui forment le chromatogramme de l’échantillon
Son temps de rétention doit être proche de celui (ou de ceux) du (ou des) soluté(s) à doser
Sa concentration doit être proche ou supérieure à celle des autres solutés pour être dans les conditions d’une réponse linéaire du détecteur
il doit être inerte vis-à-vis des composés de l’échantillon.
Rs > 1.25 (pic de l’étalon soit bien séparé des autres pics).
Voisin du pic de l’analyte
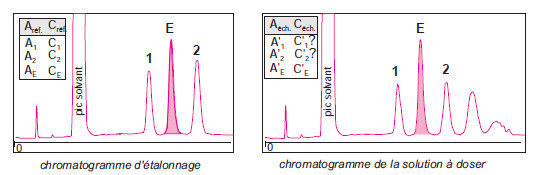
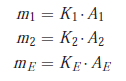
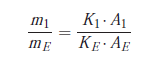
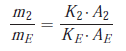
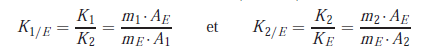
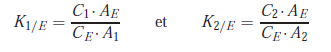
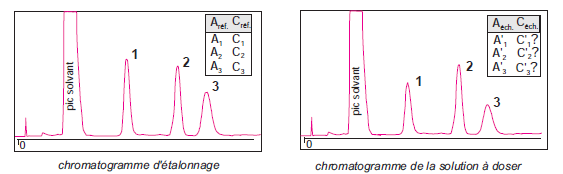
Chromatographie de l’échantillon – Calcul des concentrations
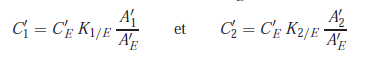
3.
MÉTHODE PAR NORMALISATION INTERNE
Cette méthode, également appelée « 100 % normalisée », est réservée aux mélanges dont on a identifié tous les constituants par autant de pics d’élution séparés sur le chromatogramme, afin de pouvoir faire le bilan complet de l’échantillon concerné.
-Permet d’éviter les erreurs associées à l’injection de l’échantillon.
-Nécessité de l’élution de tous les composants. -On mesure l’aire de tous les pics.
-Correction de ces données pour tenir compte des différences de réponse du détecteur.
-Détermination de la concentration de l’analyte à partir du rapport de cette aire à l’aire totale de tous les pics.
Inconvénient:
Difficulté de trouver les conditions pour l’élution de tous les composants du mélange dans un temps raisonnable.
1.
Calcul des coefficients de réponse relatifs
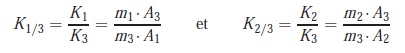
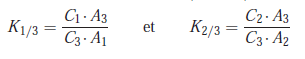
2.
Chromatographie de l’échantillon – Calcul des concentrations
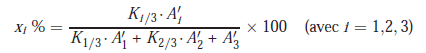
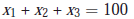
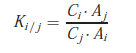
VIII.
CONCLUSION
La chromatographie est une méthode de séparation non destructrice d’un mélange liquide ou gazeux en ses différents constituants.
C’est également une méthode analytique qui a pour but d’identifier et de quantifier les composés d’un mélange.C’est une méthode de choix en analyse pharmaceutique tant biologique qu’industrielle.