
Métabolisme des acides aminés
Métabolisme des acides aminés
Parmi les 20 acides aminés protéinogènes, 8 sont indispensables à l’homme et doivent être apportés par l’alimentation.
Seuls les microorganismes et les plantes peuvent produire ces acides aminés (AA) indispensables.
La cystéine et la tyrosine sont qualifiés de semi- essentiels, car des acides aminés indispensables (Met, Phe) sont des précurseurs pour leur synthèse.
Les 10 acides aminés restants ne sont pas indispensables et peuvent être synthétisés par le corps humain (His et Arg méritent également d’être considérés comme des AA semi- indispensables, car même si notre organisme est capable de les synthétiser, c’est en quantité insuffisante pour couvrir les besoins lors de la croissance).
Alors que pour la biosynthèse des acides aminés non indispensables au plus trois enzymes sont nécessaires, la biosynthèse des acides aminés indispensables exige entre 5 (Thr) et 11 (Trp) enzyme différentes.
Les animaux supérieurs se sont manifestement limités aux synthèses les plus simples et font sous-traiter les synthèses complexes.
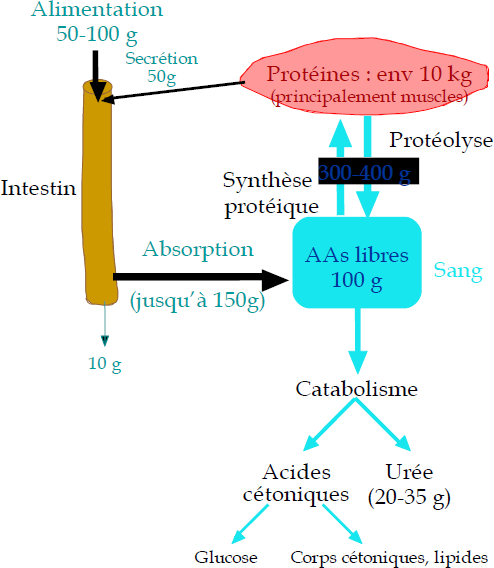
Les acides aminés parviennent normalement dans le corps à partir de l’alimentation sous forme de polymères, c’est-à-dire sous forme de protéines.
L’hydrolyse de protéines en peptides et en acides aminés isolés débute déjà dans l’estomac.
Le temps de séjour y est toutefois trop court pour une hydrolyse complète.
Des peptides plus courts parviennent ainsi dans les cellules épithéliales de l’intestin (muqueuse) grâce à des systèmes de transport et sont complètement découpés en acides aminés dans leur cytoplasme.
Les cellules de la muqueuse fournissent les acides aminés au sang, grâce auquel ils sont transportés en vue de transformations ultérieures.
Alimentation ⇒ protéines alimentaires (végétales et animales) ⇒ acides aminés (par l’action de protéases)
I-Biosynthèse des AAs
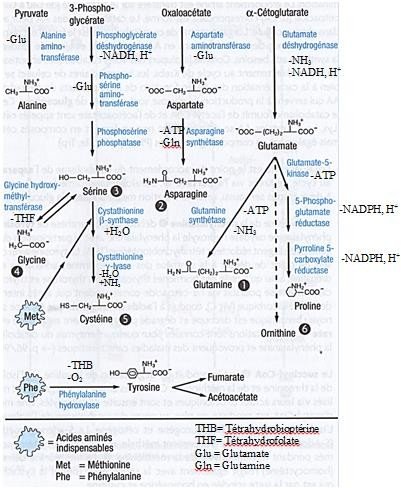
L`homme ne peut synthétiser que les AA non indispensables : Glu, Gln, Arg, Ser, Gly, Cys, Asp, Asn, Tyr, Ala, Pro.
L’alanine, l’aspartate et le glutamate jouent un rôle central dans le métabolisme des
AA.
Leur métabolisme est étroitement associé aux intermédiaires du cycle de Krebs.
Ils sont en effet produits à partir de leurs α-cétoacides respectifs, que sont le pyruvate, l’oxaloacétate et l’α-cétoglutarate, et peuvent à nouveau être convertis en ces derniers.
Il s’agit dans ce cas
de réactions réversibles de transamination.
Le glutamate est synthétisé dans les mitochondries à partir d’α-cétoglutarate et d’ammoniac libre.
Il fournit les groupements amine indispensables aux biosynthèses de l’alanine et de l’aspartate.
La proline, unique iminoacide parmi les AA protéinogènes, est synthétisé à partir du glutamate. Après phosphorylation de ce dernier, la proline est produite en trois étapes à partir de glutamate semi aldéhyde.
Les deux amides glutamine et asparagine sont produites à partir de leurs acides respectifs (Glu, Asp).
La réaction de la glutamine synthétase, qui synthétise de façon irréversible de la glutamine à partir du glutamate, sert à fixer l’azote ammoniacal produit lors du catabolisme des acides aminés.
L’asparagine est produite au cours d’une réaction de transamination, lors de laquelle le glutamate cède son groupement amine à l’aspartate.
La biosynthèse de la sérine se raccorde à un intermédiaire de la glycolyse.
La sérine est produite en trois étapes à partir de 3-phosphoglycérate.
La sérine elle-même sert de point de départ à la synthèse de la glycine et de la cystéine. Par élimination du groupement hydroxyméthyle, la sérine peut être convertie en glycine.
Cette réaction est réversible, de sorte que la sérine peut également être produite à partir de la glycine.
Dans le métabolisme général, la sérine entre dans les synthèses des phospholipides, des purines et de la cystéine.
L’arginine, dernier AA non essentiel, est produite à partir d’ornithine. Avec la
citruline, l’ornithine fait partie des deux acides aminés non protéinogènes les plus importants.
La plus grandes parti des AA formés sert à la biosynthèse des protéines.
Les ARNm servant de manuel de construction sont produits à partir de l’ADN matrice dans le noyau et sont à leur tour traduits dans le cytoplasme, au niveau des ribosomes, dans la séquence d’acides aminés de la protéine.
Pour ce faire, les AA ne se fixent pas directement aux ribosomes, mais sous la forme de leurs ARNt-aminoacylés.
A chaque AA correspond un aminoacyl-ARNt.
Remarque : indispensable aux réactions de transamination et de désamination, le coenzyme phosphate de pyridoxal (PALP) participe aux réactions importantes du métabolisme des AA.
Au cours d’une première étape il prend en charge le groupement amine, puis il le transfert à un partenaire réactionnel lors d’une deuxième étape.
Le PALP est constitué de pyridoxal qui, avec la pyridoxamine et la pyridoxine, est une des formes de la vitamine B6.
Transamination : Transfert d’un groupement aminé sur un α-céto acide Donneur de NH2 = acide glutamique ; enzyme = transaminase
Ac glutamique + α -céto acide Ac α -céto glutarique + Ac aminé
| Ac aminé | α -céto acide correspondant |
| Sérine | Ac hydroxypyruvique |
| Glutamate | Ac α -céto glutarique |
| Aspartate | Ac oxaloacétique |
| Alanine | Ac pyruvique |
Conversion : synthèse d’un AA à partir d’un autre AA Réversible : Glycine Sérine
Non réversibles :
Méthionine Cystéine par transsulfuration d’homocystéine Phénylalanine Tyrosine par hydroxylation
Aspartate Alanine par décarboxylation.
Glutamate Proline par cyclisation Autres :
Arginine : produite au cours du cycle de l’urée
AAs « non-indispensables »
Peuvent être synthétisés dans les tissus humains, mais : cette synthèse est insuffisante pour couvrir les besoins de l’organisme
Alanine : transamination de l’ac pyruvique
Asparagine : synthèse à partir de l’aspartate
Aspartate : transamination de l’ac oxaloacétique
Arginine : produite au cours du cycle de l’urée (indisp. chez l’enfant)
Cystéine : synthèse à partir de la méthionine
Glutamate : transamination de l’ac α-céto glutarique ; désamination de la glutamine
Glutamine : transfert de NH3 sur l’ac glutamique
Glycine : synthèse à partir de la sérine (interconversion) ; transamination de l’ac glyoxylique
Histidine (indisp. chez l’enfant)
Proline : synthèse à partir de l’ac glutamique
Sérine : synthèse à partir de la glycine (interconversion) transamination de l’ac phospho-3- hydroxypyruvique
Tyrosine : hydroxylation de la phénylalanine
+
Hydroxyproline et Hydroxylysine (non nécessaires à la synthèse protéique, sont formés au cours de la maturation post-traductionnelle du collagène).
II-Catabolisme des acides aminés
Le catabolisme d’un acide aminé (AA) débute en général par une réaction de transamination.
Le groupement amine est transféré sur glutamate (de Glu à Gln) et l’α- cétoacide (α-CA) correspondant est formé.
Le catabolisme des AA peut avoir lieu dans chaque cellule.
L’organe central de ce métabolisme est le foie.
Il est responsable de la constance des concentrations en AA dans le sang et ajuste le rendement de la synthèse aux besoins.
Comme la plupart des voies cataboliques aboutissent plus ou moins directement au cycle de Krebs, les intermédiaires de celui-ci se prêtent bien à la caractérisation des AA en fonction de leurs produits de dégradation.
Les AA qui servent à la production de glucose sont qualifiés de glucogènes. Ceux dont le catabolisme fournit de l’acétyl-CoA et de l’acétoacétate sont appelés cétogènes (Lys, Leu).
Certains AA se décomposent non seulement en composés cétogènes, mais également en composés glucogènes (Phe, Tyr, Thr, Ile, Trp).
Devenir du groupement NH2
Les réactions de transamination Au niveau du foie :
Les réactions de transamination, catalysées par des aminotransférases, assurent les échanges d’azote entre les acides aminés et les acides α-cétoniques : l’acide aminé, donneur du groupement amine, devient un acide α-cétonique tandis que l’acide α-cétonique accepteur devient un acide α-aminé.
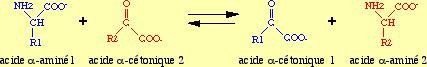
La réaction de transamination nécessite un coenzyme, le phosphate de pyridoxal (PPal), transporteur intermédiaire de la fonction amine.
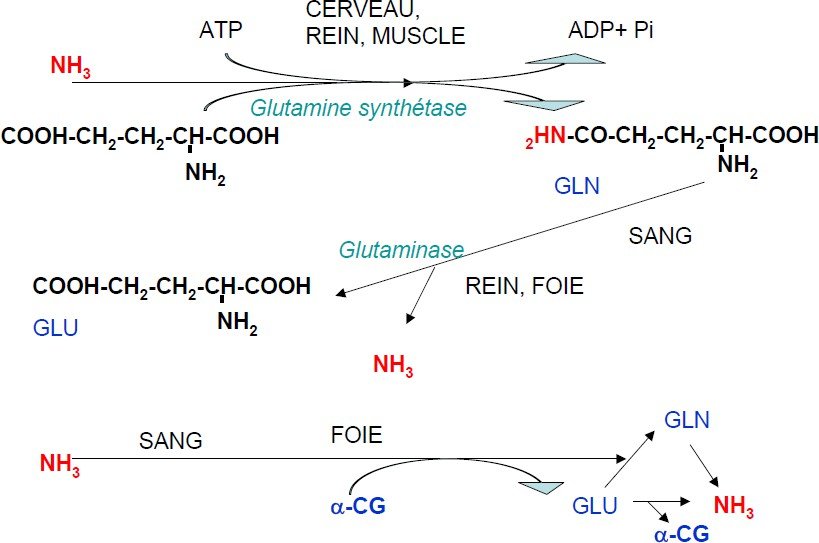
Le but est de collecter les groupements amines à partir des différents AA sous forme d`un seul d`entre eux “L glutamate´´ qui les canalisent soit vers les voies de biosynthèses soit l’excrétion sous forme d`urée.
Les cellules contiennent des transaminases différentes, spécifiques de l’acide aminé donneur.
La plupart utilise l’ α-cétoglutarate comme groupement accepteur d’amine.
Il y a, alors, production de L-glutamate.

AA + Ac α-cétoglutarique < > α -cétoacide + Ac Glutamique
La réaction de désamination du L-glutamate
Le L-glutamate, produit par les réactions de transamination, est transporté du cytosol aux mitochondries, il subit une désamination oxydative, catalysée par la L-glutamate déshydrogénase, qui éliminera finalement le groupement α-aminé sous forme de NH +.
4
Chez les mammifères, l’ion ammonium, toxique pour la cellule, sera incorporé dans le cycle de l’urée puis excrété.
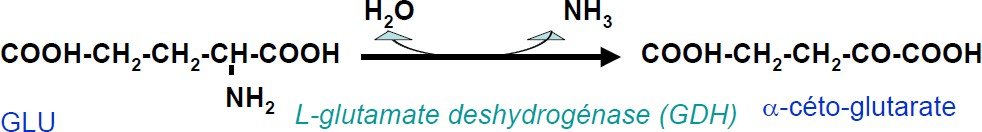
Le couplage transamination-désaminationoxydative de GLU est la voie majeure de production d’ammoniaque par les cellules.
Transport des groupements NH + des autres tissus :
3
Sous forme de Glutamine :
Le NH + sera transporté sous forme de glutamine, cette dernière peut traverser la membrane alors que le glutamate ne peut pas le faire.
3
Une fois dans le foie sous l`action d`une glutaminase au niveau des mitochondries, le groupement NH + est libéré.
3
Sous forme d’Alanine:
Au niveau du muscle les AA sont catabolisés pour fournir du carburant, les groupements NH2 seront collectés sur le Glu les réactions de transamination et le glutamate est transformé en glutamine pour ainsi passer au niveau du foie.
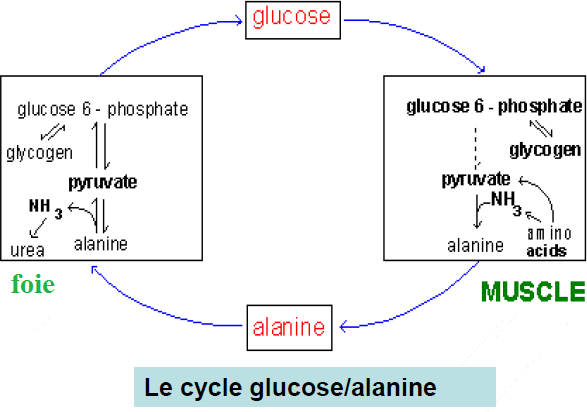
Au niveau du muscle existe une deuxième forme de transport du groupement NH2 c’est le cycle Glucose Alanine :
L’Alanine aminotransférase transfert le groupement α amine au pyruvate (produit de la glycolyse), l’Alanine formé ainsi passe dans le sang et regagne le foie ou il subit une transamination et fourni son groupement NH2 au foie et d’autre part le pyruvate qui va servir à la néoglucogenèse et de ce fait le fardeau de la néoglucogenèse du muscle est assuré par le foie épargnant ainsi de l’énergie pour le muscle.
Hyperammoniémie
Si défaut dans la Synthèse de l’Urée ⇒ NH + et AA s’accumulent dans le sang.
4
Ce phénomène se rencontre dans les maladies hépatiques graves ou lors de troubles innés du Métabolisme
La base moléculaire de cette toxicité n’est pas complètement connue.
Les étapes terminales de cette intoxication ⇒ état comateux et effets sur le cerveau⇒ Modifications du pH cellulaire⇒ Déplétion de certains intermédiaires du cycle de l’acide citrique.
Cycle de l’urée
L’ammoniac (NH3) libéré au cours du catabolisme des acides aminés doit être immédiatement transformé par l’organisme à cause de ses effets neurotoxiques.
En conditions normales, l’ammoniac (pKa 9,1) est protoné à 99% sous forme d’ion ammonium (NH +) dans le sang (pH 7,4) et dans les cellules (pH 6,0-7,1).
Sous cette forme il se dissout aisément en milieu aqueux, mais il ne peut pénétrer dans les membranes.
En cas d’augmentation du pH
4
sanguin, la réaction se déplace de NH + vers NH en faveur de l’ammoniac libre.
L’ammoniac
4 3
lui se dissout très bien dans les membranes et exerce dans ce cas son effet neurotoxique.
L’ammoniac n’est toutefois pas uniquement un poison cellulaire, il est également un élément de nombreuses biosynthèses (ADN, purines, etc.).
C’est pourquoi l’organisme des animaux supérieurs s’efforce d’épurer l’ammoniac fortement toxique en le fixant spécifiquement.
Les poissons peuvent excréter directement l’ammoniac, les mammifères l’intègrent dans l’urée et les oiseaux sont capables de le concentrer davantage en éliminant l’ammoniac sous forme d’acide urique.
Le cycle de l’urée est chez l’homme la plaque tournante de la détoxification de l’ammoniac.
Il se déroule dans le foie.
C’est là que s’accumule le glutamate, en tant que produit final des différentes voies cataboliques des AA.
De l’ammoniac est libéré dans la mitochondrie au cours des réactions de désamination.
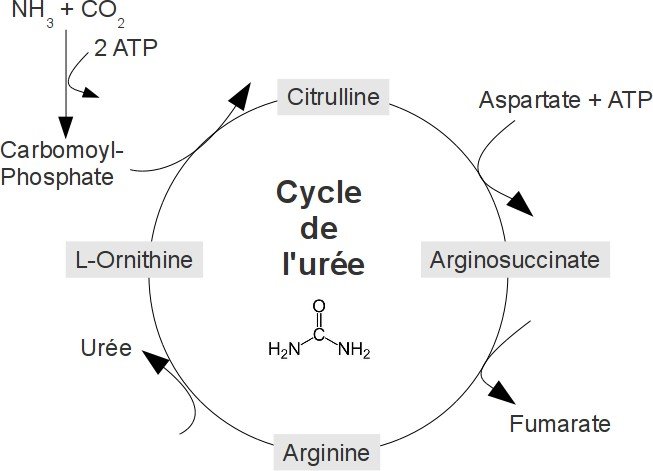
Sous la forme d’ion d’ammonium, il se condense avec l’hydrogénocarbonate pour donner du carbamoyl phosphate. Pour synthétiser
l’urée, qui contient deux atomes de d’azote, un autre atome d’azote doit encore entrer dans le cycle réactionnel.
Celui-ci provient de l’aspartate, qui est lui-même obtenu par transamination à partir de glutamate et d’oxaloacétate.
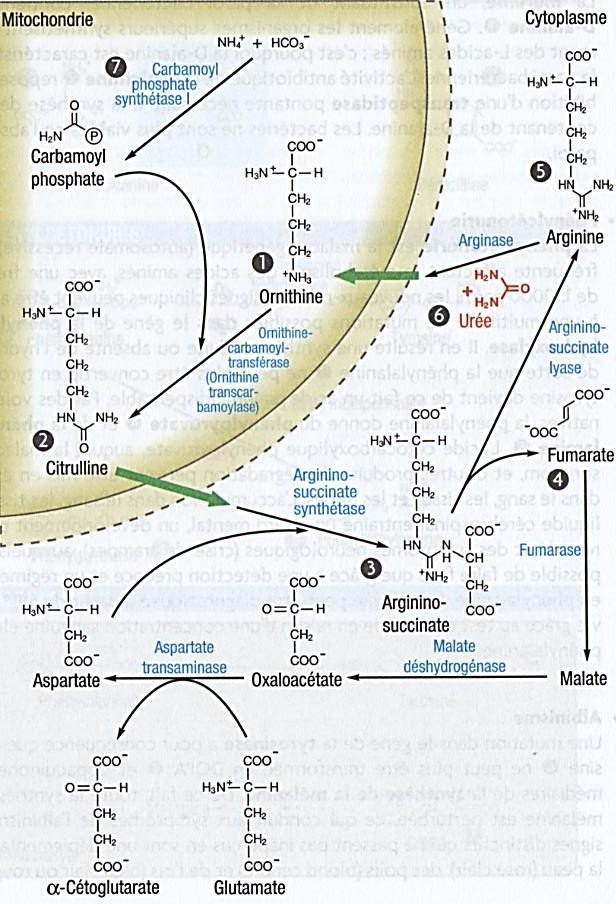
L’acide aminé non protéinogène ornithine(1) réagit avec le carbamoyl phosphate formé à partir du glutamate pour donner de la citrulline(2).
La citrulline est transportée depuis la mitochondrie vers le cytoplasme et y forme avec l’aspartate de l’argininosuccinate(3). L’argininosuccinate lyase en libère du fumarate(4), qui redonne de l’oxaloacétate par transamination.
Il en résulte l’acide aminé protéinogène arginine(5).
Celui-ci peut entrer dans la synthèse protéique ou être transformé en ornithine par l’arginase en libérant de l’urée(6).
Dès que l’ornithine regagne la mitochondrie par transport actif, la séquence réactionnelle du cycle de l’urée est achevée.
En tout cinq réactions enzymatiques sont impliquées dans la biosynthèse de l’urée.
Deux d’entre-elles se déroulent dans la mitochondrie et trois dans le cytoplasme.
La carbamoyl phosphate synthétase (CPS) est l’enzyme-clé qui régule le cycle de l’urée.
Deux isoformes de cette enzyme sont distinguées.
L’isoforme CPS I(7) est mitochondriale et intervient dans le cycle de l’urée.
Au contraire, l’isoforme CPS II est cytoplasmique et intervient dans la synthèse des pyrimidines.
En outre, elles se différencient dans le choix du substrat, CPS I condense l’ion ammonium et l’ion hydrogénocarbonate et CPS II a la glutamine comme substrat.
De façon physiologiquement utile, la biosynthèse de l’urée est reliée à la concentration en acides aminés dans le sang portal qui circule vers le foie.
Il existe une relation approximativement linéaire entre la concentration en azote dans le plasma et la synthèse de l’urée.
La CPS I réagit très vite aux variations de concentration en N-acétyl-glutamate.
Si cette substance-signal vient à manquer, la CPS I est alors totalement inactivée.
Pour synthétiser du N-acétyl-glutamate, il faut une augmentation de la concentration sanguine en glutamate obtenue en cas d’apport protéique.
Régulation de la biosynthèse de l’urée :
| Inhibée par | Activée par | |
| Carbamoyl phosphate synthase I | Concentration plasmatiqueen azote aminé< 4,5 mg / 100 ml | Augmentation de laconcentration plasmatique en azote aminé |
| N-acétyl-glutamate | N-acétyl-glutamate |
Bilan énergétique
CO2 + NH + + 3 ATP + Asp + 2 H O——> URÉE + 2 ADP + 2 P + AMP + PP + fumarate
4
2
i
i
un atome d’azote de l’urée provient de l’ammoniac, l’autre de l’aspartate
presque tous les acides aminés servent de donneurs de groupe aminé en formant du glutamate par transamination
chez l’homme, il est extrêmement important qu’il y ait un apport d’arginine par l’alimentation.
En effet, la quantité qu’il est capable de synthétiser est insuffisante pour entrer dans la composition des protéines et servir à la production d’urée
enfin, si l’on revient au cycle général du devenir de l’azote, celui-ci est rejeté dans la biosphère après dénitrification par des bactéries
II-2- Devenir du groupement carboxylique :
Les AA peuvent subir une décarboxylation ⇒Amines
Amines ⇒ Aldehydes(R-CHO) + NH3+ (mono amino oxydase) His ⇒ histamine Trp ⇒ tryptamine
Tyr ⇒ tyramine Cys ⇒ cysteamine.
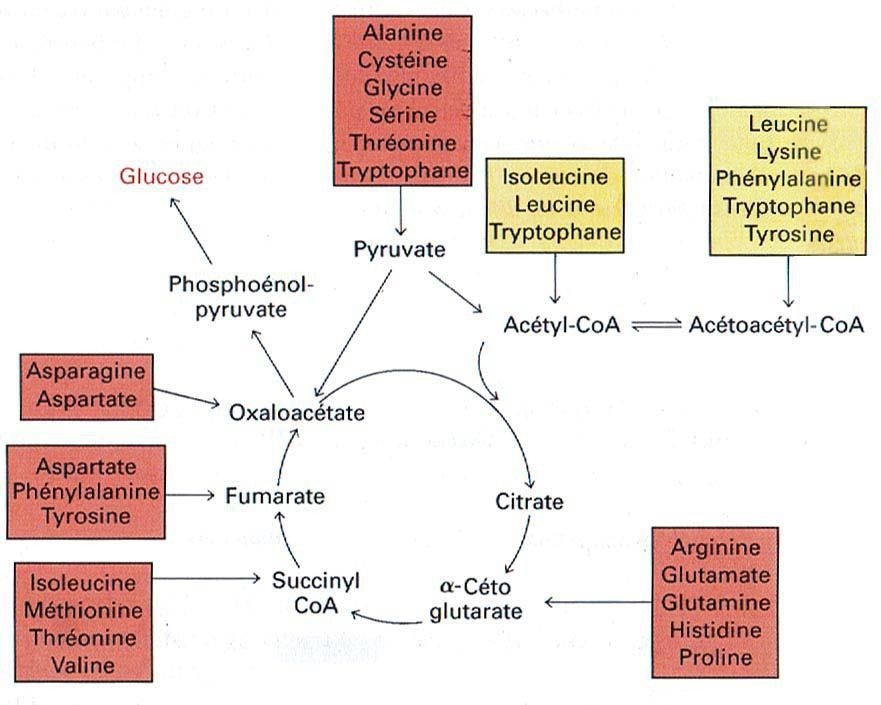
Les caractéristiques principales de la dégradation du squelette carboné sont les suivantes :
Cette dégradation est un exemple remarquable d’économie des transformations métaboliques.
En effet, le catabolisme de l’ensemble des squelettes carbonésdes 20 acides aminés est canalisé en seulement 7 molécules :
le pyruvate
des intermédiaires du cycle de Krebs (oxaloacétate, α-cétoglutarate, succinyl CoA et fumarate)
l’acétyl CoAou l’acéto-acétyl CoA
Les acides aminés dégradés en acétyl CoA, en acéto-acétyl CoA ou en d’autres dérivés du coenzyme A, sont dits cétogènes puisqu’ils contribuent à former des corps cétoniques.
Ceux qui aboutissent au pyruvate ou à un intermédiaire du cycle de Krebs sont dits glucogènes.
En effet, ils peuvent être convertis en phosphoénolpyruvate et alimenter la voie de la néoglucogénèse.
Cette classification n’est pas absolue puisqu’elle dépend du devenir réel d’un métabolite considéré.
C- Quelques exemples caractéristiques du catabolisme du squelette carboné :
L’oxaloacétate est le point de raccordement du catabolisme de l’asparagine(1) au cycle de Krebs via l’aspartate(2). L’oxaloacétate formé lors d’une réaction à deux étapes sert aussi, selon la situation métabolique, à la gluconéogenèse.
Le catabolisme de la phénylalanine(3) débute avec la synthèse de la tyrosine(4).
La phénylalanine oxydase hydrolyse la phénylalanine avec la participation du dioxygène et de l’agent réducteur tétrahydrobioptérine (THB).
La tyrosine ainsi obtenue sert de point de départ à la synthèse des catécholamines adrénaline et noradrénaline, ainsi qu’à celle des hormones thyroïdiennes thyronine et thyroxine.
Le catabolisme se poursuit via l’α-cétoacide correspondant qui, après intervention de l’acide ascorbique (vit C), conduit à l’acide homogentisique(5). A ce stade, le noyau benzénique est découpé et dégradé progressivement en acétate et fumarate(6). Des mutations sont connues pour quelques enzymes du catabolisme de la phénylalanine et provoquent des maladies caractéristiques.
Le succinyl-CoA(7) est le produit de dégradation de la valine, de l’isoleucine, de la thréonine et de la méthionine.
Ces acides aminés sont également métabolisés via leurs acides α-cétoniques et sont ensuite décarboxylés.
Une molécule d’acétyl-CoA est produite en plus au cours du catabolisme de l’isoleucine(8).
L’isoleucine est de ce fait glucogène et cétogène.
La S-adénosylméthionine (S-AdoMet), donneur de groupement méthyle, et l’AA soufré cystéine sont formés pendant le catabolisme de la méthionine(9).
La méthionine déméthylée (homocystéine) réagit également avec la sérine pour former la cystathionine, qui est par la suite scindée en homosérine et cystéine.

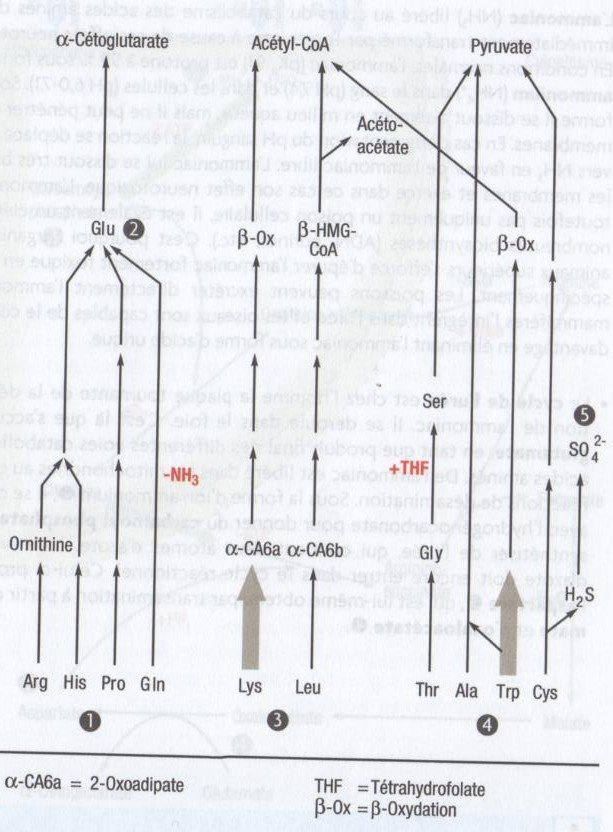
Le catabolisme de l’arginine, de l’histidine, de la proline et de la glutamine(1) conduit au glutamate(2). La transamination du glutamate produit l’intermédiaire α- cétoglutarate.
Le glutamate a une importance primordiale dans le métabolisme des AA : d’une part parce qu’il intervient dans le métabolisme de l’ammoniac, d’autre part parce qu’il sert de donneur de groupement amine dans les réactions de transamination.
Les AA lysine et leucine(3) sont catabolisés en acétyl-CoA.
Dans le cas de la lysine, plusieurs voies conduisent d’abord à l’α-cétoacide correspondant.
Celui-ci est dégradé en trois étapes en crotonyl-CoA, composé qui parcourt ensuite la β-oxydation qui s’achève avec l’acétyl-CoA.
La leucine, comme les deux autres acides aminés à chaîne ramifiée (Val, Ile), est dégradée par un complexe déshydrogénase après transamination en α-cétoacide.
Une mutation au niveau de cette enzyme conduit à la maladie du sirop d’érable.
Du β-hydroxy-β- méthylglutaryl-CoA (β-HMG-CoA) est produit avant la fin de la voie métabolique.
De l’acétoacétate et de l’acétyl-CoA sont ensuite formés à partir du β-HMG-CoA.
Le pyruvate est le produit de dégradation de la thréonine, du tryptophane, de l’alanine, de la sérine, de la glycine et de la cystéine(4). La thréonine est catabolisée en pyruvate en trois étapes via la glycine et la sérine.
Le catabolisme de l’alanine en pyruvate fait
intervenir une unique réaction de transamination, étant donné que leur squelette carboné est identique. le tryptophane est transformé en plusieurs étapes en acide α-cétoadipique.
De l’alanine est produite par clivage au cours de ces réactions ; elle donnera du pyruvate.
Le reste du squelette du tryptophane est transformé en acétyl-CoA par la β-oxydation.
Le tryptophane revêt une importance particulière en tant que précurseur de la nicotinamide (la vitamine B3) et de la sérotonine (une hormone tissulaire).
Lors du catabolisme de la cystéine, outre le produit final qu’est le pyruvate, des ions sulfate(5) sont aussi libérés.
Une alimentation riche en protéines acidifie de ce fait les urines.
Clinique
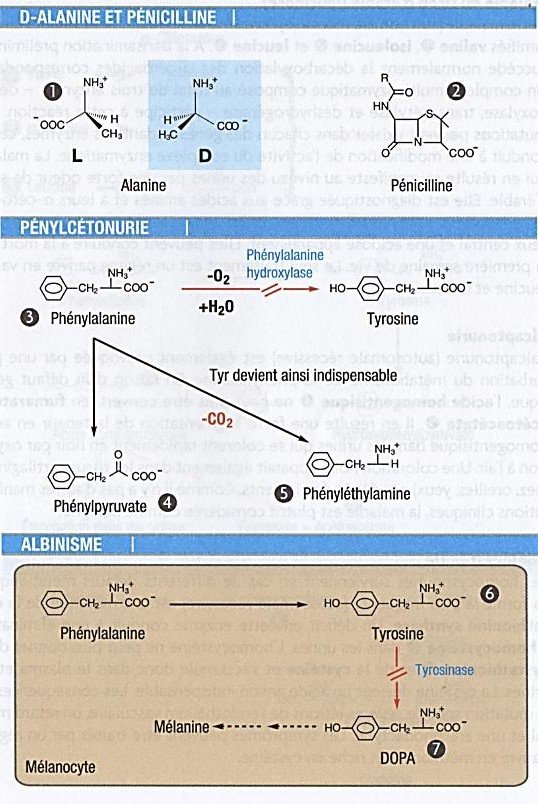
D-alanine et pénicilline
La muréine, un constituant de la paroi bactérienne, contient de la D-alanine(1). Généralement les organismes supérieurs synthétisent uniquement des L-acides aminés ; c’est pourquoi la D-alanine est caractéristique de la paroi bactérienne.
L’activité antibiotique de la pénicilline(2) repose sur l’inhibition d’une transpeptidase pontante nécessaire à la synthèse des parois contenant de la D-alanine.
Les bactéries ne sont plus viables en l’absence de paroi.
Phénylcétonurie
La phénylcétonurie est la maladie génétique (autosomale récessive) la plus fréquente affectant le métabolisme des acides aminés, avec une fréquence de 1 :10000 parmi les nouveau-nés.
Les signes cliniques peuvent être attribués à une multitude de mutations possibles dans le gène de la phénylalanine hydroxylase.
Il en résulte une synthèse réduite ou absente de l’hydroxylase, de sorte que la phénylalanine(3) ne peut plus être convertie en tyrosine.
La tyrosine devient de ce fait un acide aminé indispensable.
Par des voies alternatives, la phénylalanine donne du phénylpyruvate(4) et de la phényléthylamine(5).
L’acide cétocarboxylique phénylpyruvate, auquel la maladie doit son nom, et d’autres produits de dégradation peuvent être mis en évidence dans le sang, les tissus et les urines.
L’accumulation dans le sang, les tissus et le liquide cérébrospinal entraîne un retard mental, un développement corporel retardé et des symptômes neurologiques (crise de crampes), auxquels il n’est possible de faire face que grâce à une détection précoce et un régime pauvre en phénylalanine.
La maladie peut être diagnostiquée à partir du 5ème jour de
vie grâce au test de Guthrie en raison d’une concentration sanguine élevée en phénylalanine.
Albinisme
Une mutation dans le gène de la tyrosinase a pour conséquence que la tyrosine(6) ne peut plus être transformée en DOPA(7) et dopamine, intermédiaires de la synthèse de la mélanine.
De ce fait, toute la synthèse de la mélanine est perturbée, ce qui conduit aux symptômes de l’albinisme.
Des signes distinctifs qui ne passent pas inaperçus en sont une dépigmentation de la peau (rose clair), des poils (blond cendré) et de l’iris (bleu clair ou rougeâtre).
Maladie du sirop d’érable (leucinose)
Cette maladie (autosomale récessive) concerne la dégradation des acides aminés ramifiés valine(1), isoleucine(2) et leucine(3).
A la transamination préliminaire succède normalement la décarboxylation des α-cétoacides correspondants.
Un complexe multienzymatique composé au total de trois enzymes – décarboxylase, transacétylase et déshydrogénase- participe à cette réaction.
Des mutations peuvent exister dans chacun des gènes codant ces enzymes, ce qui conduit à une modification de l’activité du complexe enzymatique.
La maladie qui en résulte se manifeste au niveau des urines par une forte
odeur de sirop d’érable.
Elle est diagnostiquée grâce aux AA et leurs α-céto- ou α- hydroxyacides présent dans le sang et les urines.
Des lésions du système nerveux central et une acidose apparaissent.
Elles peuvent conduire à la mort dès la première semaine de vie.
Seul traitement est un régime pauvre en valine, leucine est isoleucine.
Alcaptonurie
L’alcaptonurie (autosomale récessive) est également provoquée par une perturbation du métabolisme de la phénylalanine.
En raison d’un défaut génétique, l’acide homogentisique(4) ne peut plus être converti en fumarate et acétoacétate(5).
Il en résulte une forte augmentation de la teneur en acide homogentisique dans les urines qui se colorent rapidement en noir par oxydation à l’air.
Une coloration noire apparaît également dans les tissus cartilagineux (nez, oreilles, yeux) par dépôt de pigments.
Comme il n’y a pas d’autres manifestations cliniques, la maladie est plutôt considérée comme bénigne.
Homocystinurie
Les homocystinuries surviennent en cas de différents défauts métaboliques.
La forme la plus fréquente (autosomale récessive) est une mutation de la cystathionine synthase.
Un déficit en cette enzyme conduit à une élimination d’homocystéine(6) dans les urines.
L’homocystéine ne peut plus donner de la cystathionine(7) et de la cystéine et s’accumule donc dans le plasma et les urines.
La cystéine devient un AA indispensable.
Les conséquences de la mutation sont sévères lésions de l’endothélium vasculaire, un retard mental et une arachnodactylie.
Les symptômes peuvent être traités par régime pauvre en méthionine et riche en cystéine.